

Frameshift Variant in AMPD2 in Cirneco dell'Etna Dogs with Retinopathy and Tremors
- PMID: 38397227
- PMCID: PMC10887799
- DOI: 10.3390/genes15020238
Frameshift Variant in AMPD2 in Cirneco dell'Etna Dogs with Retinopathy and Tremors
Abstract
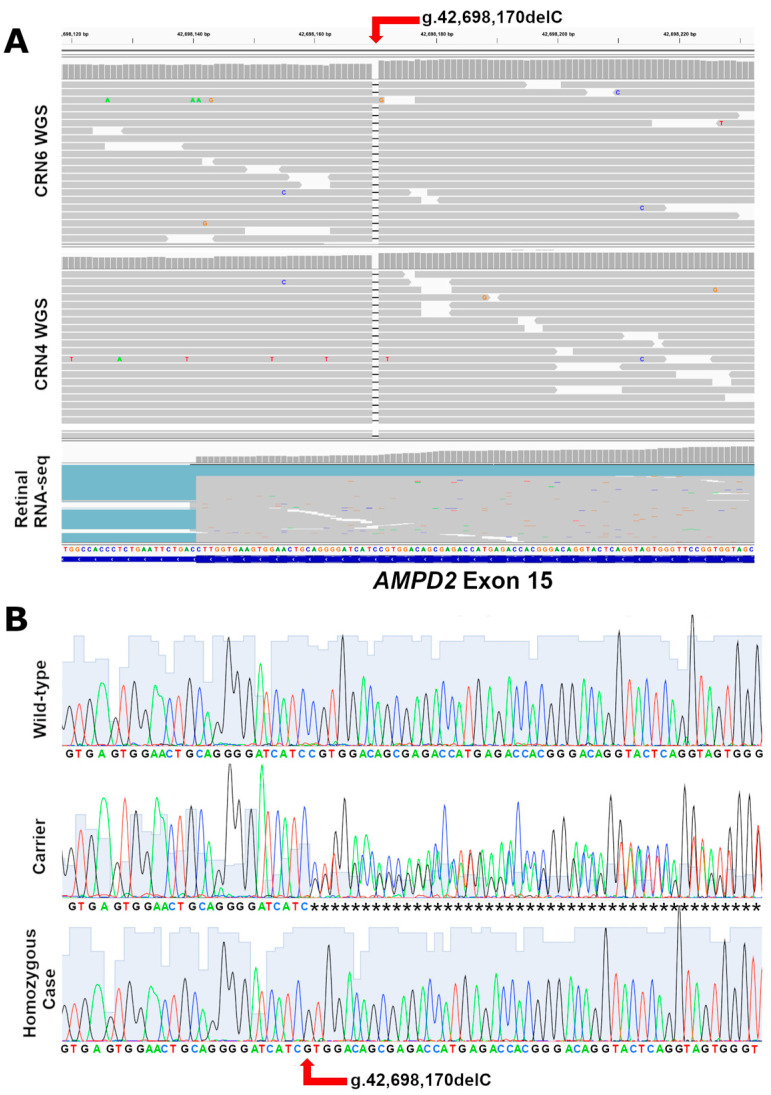
While the manifestations of many inherited retinal disorders are limited to loss of vision, others are part of a syndrome that affects multiple tissues, particularly the nervous system. Most syndromic retinal disorders are thought to be recessively inherited. Two dogs out of a litter of Cirneco dell' Etna dogs, both males, showed signs of retinal degeneration, along with tremors and signs described as either atypical seizures or paroxysmal dyskinesias, while the other two male littermates were normal. We named this oculo-neurological syndrome CONS (Cirneco oculo-neurological syndrome), and undertook homozygosity mapping and whole-genome sequencing to determine its potential genetic etiology. Notably, we detected a 1-bp deletion in chromosome 6 that was predicted to cause a frameshift and premature stop codon within the canine AMPD2 gene, which encodes adenosine monophosphate deaminase, an enzyme that converts adenosine 5'-monophosphate (AMP) to inosine 5'-monophosphate (IMP). Genotyping of the available Cirneco population suggested perfect segregation between cases and controls for the variant. Moreover, this variant was absent in canine genomic databases comprised of thousands of unaffected dogs. The AMPD2 genetic variant we identified in dogs presents with retinal manifestations, adding to the spectrum of neurological manifestations associated with AMPD2 variants in humans.
Keywords: animal model; inherited canine disease; oculo-neurological syndrome; syndromic retinal condition.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures




Similar articles
-
CCDC66 frameshift variant associated with a new form of early-onset progressive retinal atrophy in Portuguese Water Dogs.Sci Rep. 2020 Dec 3;10(1):21162. doi: 10.1038/s41598-020-77980-5. Sci Rep. 2020. PMID: 33273526 Free PMC article.
-
Complex Structural PPT1 Variant Associated with Non-syndromic Canine Retinal Degeneration.G3 (Bethesda). 2019 Feb 7;9(2):425-437. doi: 10.1534/g3.118.200859. G3 (Bethesda). 2019. PMID: 30541930 Free PMC article.
-
A LINE-1 insertion situated in the promoter of IMPG2 is associated with autosomal recessive progressive retinal atrophy in Lhasa Apso dogs.BMC Genet. 2020 Sep 7;21(1):100. doi: 10.1186/s12863-020-00911-w. BMC Genet. 2020. PMID: 32894063 Free PMC article.
-
Advances in the molecular understanding of canine retinal diseases.J Small Anim Pract. 2005 Aug;46(8):371-80. doi: 10.1111/j.1748-5827.2005.tb00333.x. J Small Anim Pract. 2005. PMID: 16119056 Review.
-
Natural models for retinitis pigmentosa: progressive retinal atrophy in dog breeds.Hum Genet. 2019 May;138(5):441-453. doi: 10.1007/s00439-019-01999-6. Epub 2019 Mar 23. Hum Genet. 2019. PMID: 30904946 Review.
Cited by
-
GTPBP2 in-frame deletion in canine model with non-syndromic progressive retinal atrophy.Sci Rep. 2025 Feb 19;15(1):6079. doi: 10.1038/s41598-025-89446-7. Sci Rep. 2025. PMID: 39971978 Free PMC article.
References
-
- Thiadens A.A., Phan T.M.L., Zekveld-Vroon R.C., Leroy B.P., van den Born L.I., Hoyng C.B., Klaver C.C., Writing Committee for the Cone Disorders Study Group Consortium. Roosing S., Pott J.-W.R., et al. Clinical Course, Genetic Etiology, and Visual Outcome in Cone and Cone–Rod Dystrophy. Ophthalmology. 2012;119:819–826. doi: 10.1016/j.ophtha.2011.10.011. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
