

Robust Approaches to the Quantitative Analysis of Genome Formula Variation in Multipartite and Segmented Viruses
- PMID: 38400045
- PMCID: PMC10892338
- DOI: 10.3390/v16020270
Robust Approaches to the Quantitative Analysis of Genome Formula Variation in Multipartite and Segmented Viruses
Abstract
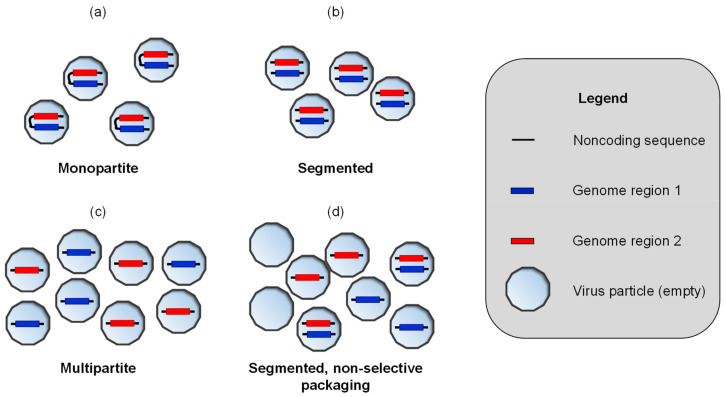
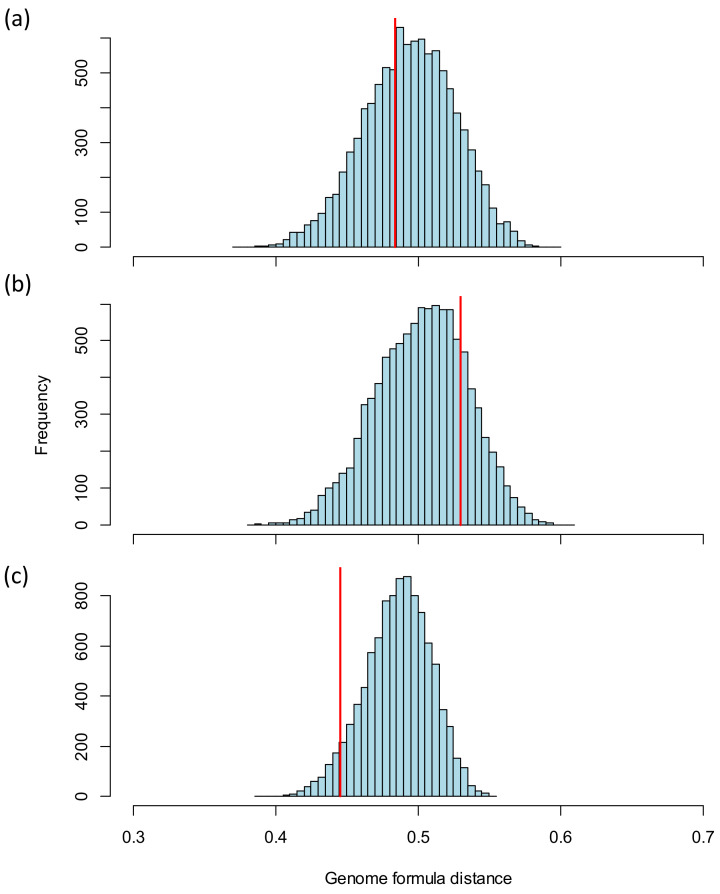
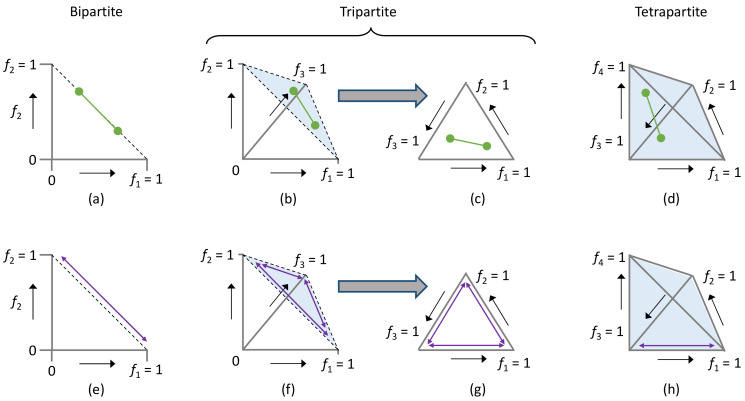
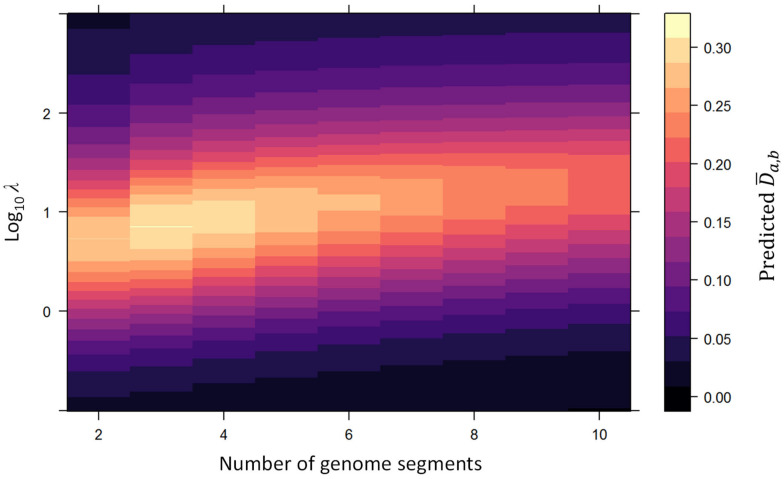
When viruses have segmented genomes, the set of frequencies describing the abundance of segments is called the genome formula. The genome formula is often unbalanced and highly variable for both segmented and multipartite viruses. A growing number of studies are quantifying the genome formula to measure its effects on infection and to consider its ecological and evolutionary implications. Different approaches have been reported for analyzing genome formula data, including qualitative description, applying standard statistical tests such as ANOVA, and customized analyses. However, these approaches have different shortcomings, and test assumptions are often unmet, potentially leading to erroneous conclusions. Here, we address these challenges, leading to a threefold contribution. First, we propose a simple metric for analyzing genome formula variation: the genome formula distance. We describe the properties of this metric and provide a framework for understanding metric values. Second, we explain how this metric can be applied for different purposes, including testing for genome-formula differences and comparing observations to a reference genome formula value. Third, we re-analyze published data to illustrate the applications and weigh the evidence for previous conclusions. Our re-analysis of published datasets confirms many previous results but also provides evidence that the genome formula can be carried over from the inoculum to the virus population in a host. The simple procedures we propose contribute to the robust and accessible analysis of genome-formula data.
Keywords: RT-PCR; genome formula; multipartite virus; plant virus; segmented virus; sequencing; statistical analysis; virus ecology; virus evolution.
Conflict of interest statement
The authors declare no conflicts of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
Figures
Similar articles
-
Evolutionary dynamics of genome segmentation in multipartite viruses.Proc Biol Sci. 2012 Sep 22;279(1743):3812-9. doi: 10.1098/rspb.2012.1086. Epub 2012 Jul 4. Proc Biol Sci. 2012. PMID: 22764164 Free PMC article.
-
The Genome Segments of Bluetongue Virus Differ in Copy Number in a Host-Specific Manner.J Virol. 2020 Dec 9;95(1):e01834-20. doi: 10.1128/JVI.01834-20. Print 2020 Dec 9. J Virol. 2020. PMID: 33028716 Free PMC article.
-
Theoretical approaches to disclosing the emergence and adaptive advantages of multipartite viruses.Curr Opin Virol. 2018 Dec;33:89-95. doi: 10.1016/j.coviro.2018.07.018. Epub 2018 Aug 16. Curr Opin Virol. 2018. PMID: 30121469 Review.
-
Cheating leads to the evolution of multipartite viruses.PLoS Biol. 2023 Apr 24;21(4):e3002092. doi: 10.1371/journal.pbio.3002092. eCollection 2023 Apr. PLoS Biol. 2023. PMID: 37093882 Free PMC article.
-
The Curious Strategy of Multipartite Viruses.Annu Rev Virol. 2020 Sep 29;7(1):203-218. doi: 10.1146/annurev-virology-010220-063346. Annu Rev Virol. 2020. PMID: 32991271 Review.
References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
