

High-throughput drug target discovery using a fully automated proteomics sample preparation platform
- PMID: 38404368
- PMCID: PMC10882491
- DOI: 10.1039/d3sc05937e
High-throughput drug target discovery using a fully automated proteomics sample preparation platform
Abstract
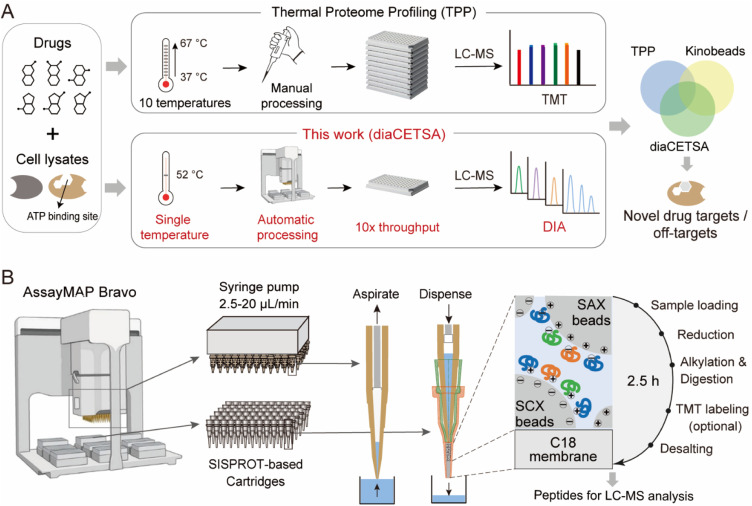
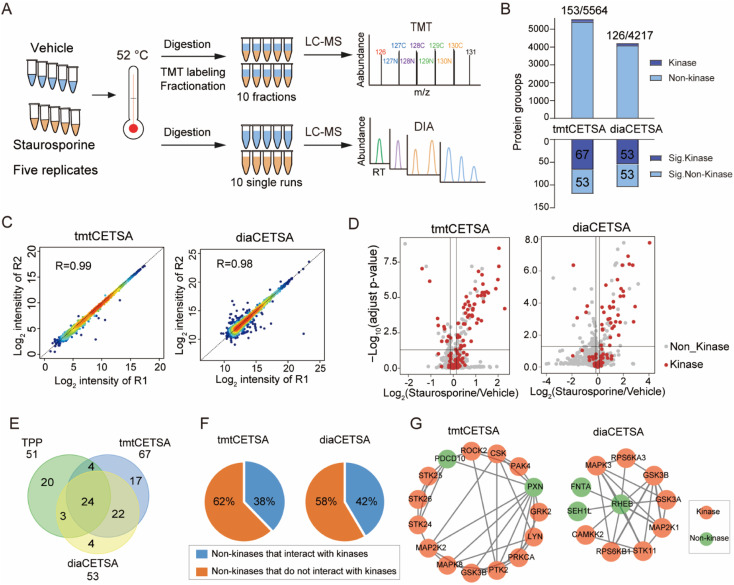
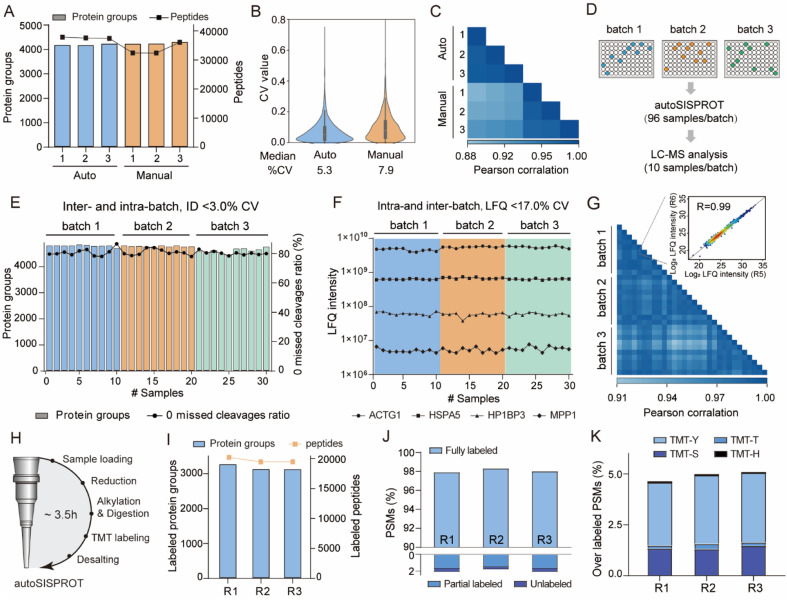
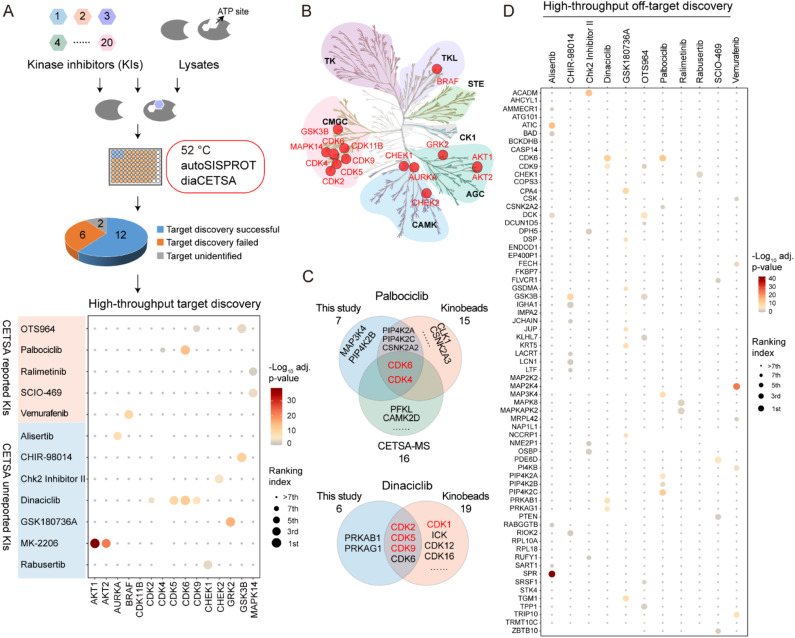
Drug development is plagued by inefficiency and high costs due to issues such as inadequate drug efficacy and unexpected toxicity. Mass spectrometry (MS)-based proteomics, particularly isobaric quantitative proteomics, offers a solution to unveil resistance mechanisms and unforeseen side effects related to off-targeting pathways. Thermal proteome profiling (TPP) has gained popularity for drug target identification at the proteome scale. However, it involves experiments with multiple temperature points, resulting in numerous samples and considerable variability in large-scale TPP analysis. We propose a high-throughput drug target discovery workflow that integrates single-temperature TPP, a fully automated proteomics sample preparation platform (autoSISPROT), and data independent acquisition (DIA) quantification. The autoSISPROT platform enables the simultaneous processing of 96 samples in less than 2.5 hours, achieving protein digestion, desalting, and optional TMT labeling (requires an additional 1 hour) with 96-channel all-in-tip operations. The results demonstrated excellent sample preparation performance with >94% digestion efficiency, >98% TMT labeling efficiency, and >0.9 intra- and inter-batch Pearson correlation coefficients. By automatically processing 87 samples, we identified both known targets and potential off-targets of 20 kinase inhibitors, affording over a 10-fold improvement in throughput compared to classical TPP. This fully automated workflow offers a high-throughput solution for proteomics sample preparation and drug target/off-target identification.
This journal is © The Royal Society of Chemistry.
Conflict of interest statement
R. T. is a founder of BayOmics, Inc. The other authors declare no competing interests.
Figures
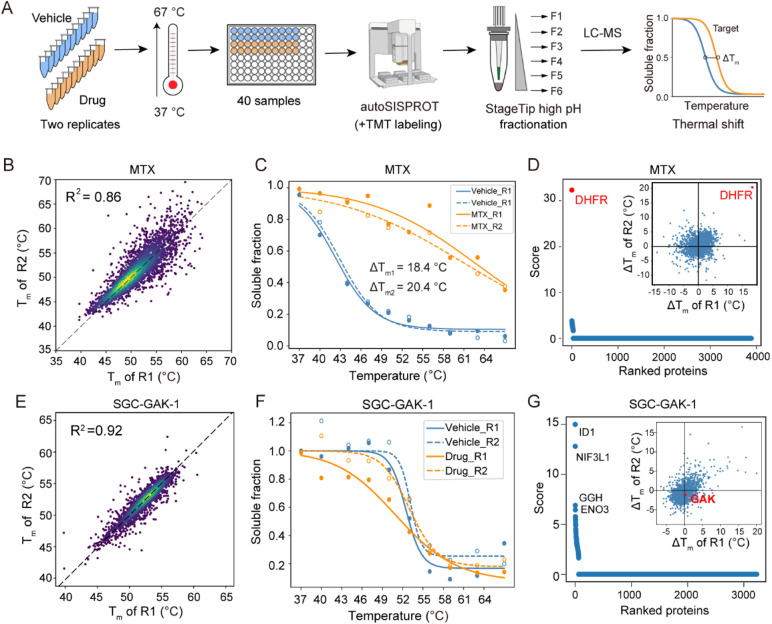
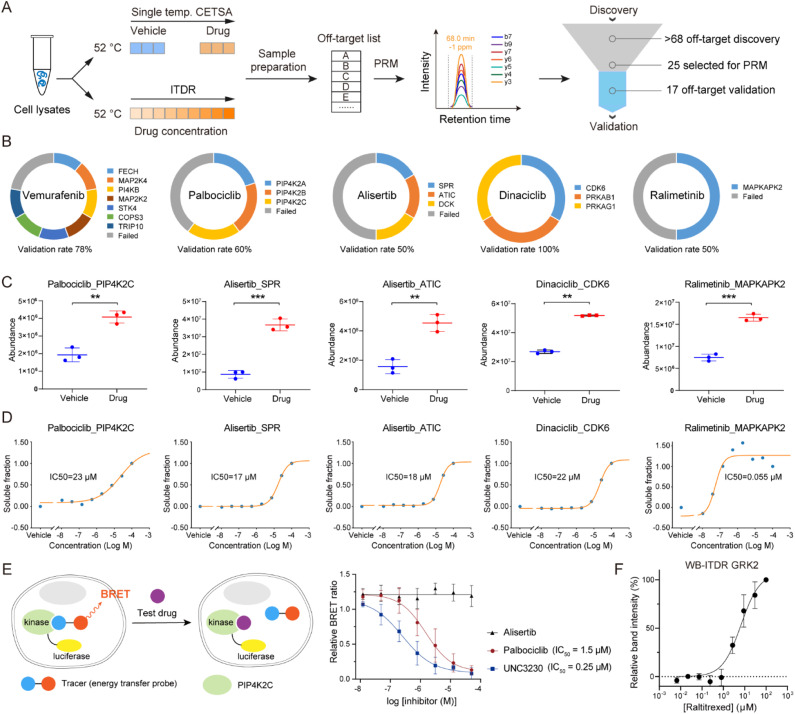
References
-
- Zecha J. Bayer F. P. Wiechmann S. Woortman J. Berner N. Müller J. Schneider A. Kramer K. Abril-Gil M. Hopf T. Reichart L. Chen L. Hansen F. M. Lechner S. Samaras P. Eckert S. Lautenbacher L. Reinecke M. Hamood F. Prokofeva P. Vornholz L. Falcomatà C. Dorsch M. Schröder A. Venhuizen A. Wilhelm S. Médard G. Stoehr G. Ruland J. Grüner B. M. Saur D. Buchner M. Ruprecht B. Hahne H. The M. Wilhelm M. Kuster B. Science. 2023;380:93–101. doi: 10.1126/science.ade3925. - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources