

This is a preprint.
Joint, multifaceted genomic analysis enables diagnosis of diverse, ultra-rare monogenic presentations
- PMID: 38405764
- PMCID: PMC10888768
- DOI: 10.1101/2024.02.13.580158
Joint, multifaceted genomic analysis enables diagnosis of diverse, ultra-rare monogenic presentations
Update in
-
Joint, multifaceted genomic analysis enables diagnosis of diverse, ultra-rare monogenic presentations.Nat Commun. 2025 Aug 7;16(1):7267. doi: 10.1038/s41467-025-61712-2. Nat Commun. 2025. PMID: 40770127 Free PMC article.
Abstract
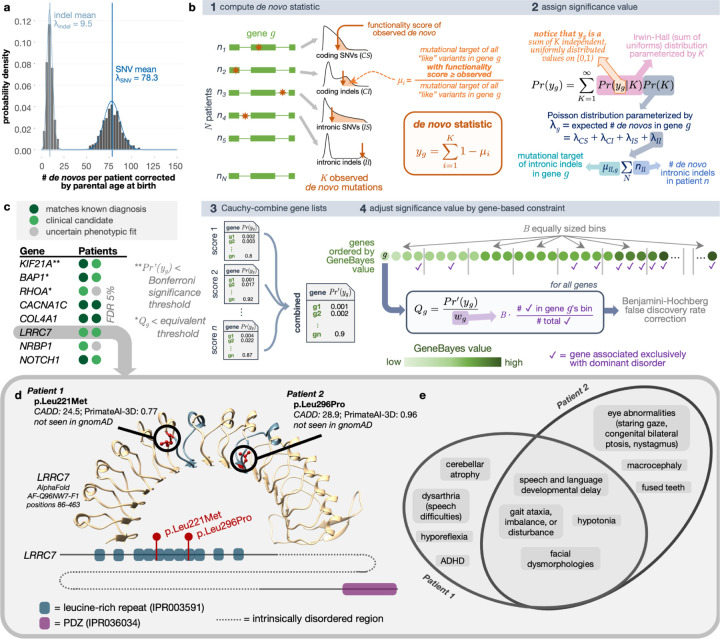
Genomics for rare disease diagnosis has advanced at a rapid pace due to our ability to perform "N-of-1" analyses on individual patients with ultra-rare diseases. The increasing sizes of ultra-rare disease cohorts internationally newly enables cohort-wide analyses for new discoveries, but well-calibrated statistical genetics approaches for jointly analyzing these patients are still under development.1,2 The Undiagnosed Diseases Network (UDN) brings multiple clinical, research and experimental centers under the same umbrella across the United States to facilitate and scale N-of-1 analyses. Here, we present the first joint analysis of whole genome sequencing data of UDN patients across the network. We introduce new, well-calibrated statistical methods for prioritizing disease genes with de novo recurrence and compound heterozygosity. We also detect pathways enriched with candidate and known diagnostic genes. Our computational analysis, coupled with a systematic clinical review, recapitulated known diagnoses and revealed new disease associations. We further release a software package, RaMeDiES, enabling automated cross-analysis of deidentified sequenced cohorts for new diagnostic and research discoveries. Gene-level findings and variant-level information across the cohort are available in a public-facing browser (https://dbmi-bgm.github.io/udn-browser/). These results show that N-of-1 efforts should be supplemented by a joint genomic analysis across cohorts.
Figures



References
-
- Marx J. L. The cystic fibrosis gene is found. Science 245, 923–925 (1989). - PubMed
Publication types
Grants and funding
- U01 HG012009/HG/NHGRI NIH HHS/United States
- U01 NS134356/NS/NINDS NIH HHS/United States
- R01 HG012286/HG/NHGRI NIH HHS/United States
- KL2 TR002552/TR/NCATS NIH HHS/United States
- R01 HL164409/HL/NHLBI NIH HHS/United States
- R35 GM127131/GM/NIGMS NIH HHS/United States
- T32 GM007748/GM/NIGMS NIH HHS/United States
- R21 HG010391/HG/NHGRI NIH HHS/United States
- U2C NS132415/NS/NINDS NIH HHS/United States
- U01 HG007530/HG/NHGRI NIH HHS/United States
- F32 HG000130/HG/NHGRI NIH HHS/United States
- R01 MH101244/MH/NIMH NIH HHS/United States
LinkOut - more resources
Full Text Sources