

Low-input and single-cell methods for Infinium DNA methylation BeadChips
- PMID: 38407446
- PMCID: PMC11040145
- DOI: 10.1093/nar/gkae127
Low-input and single-cell methods for Infinium DNA methylation BeadChips
Abstract
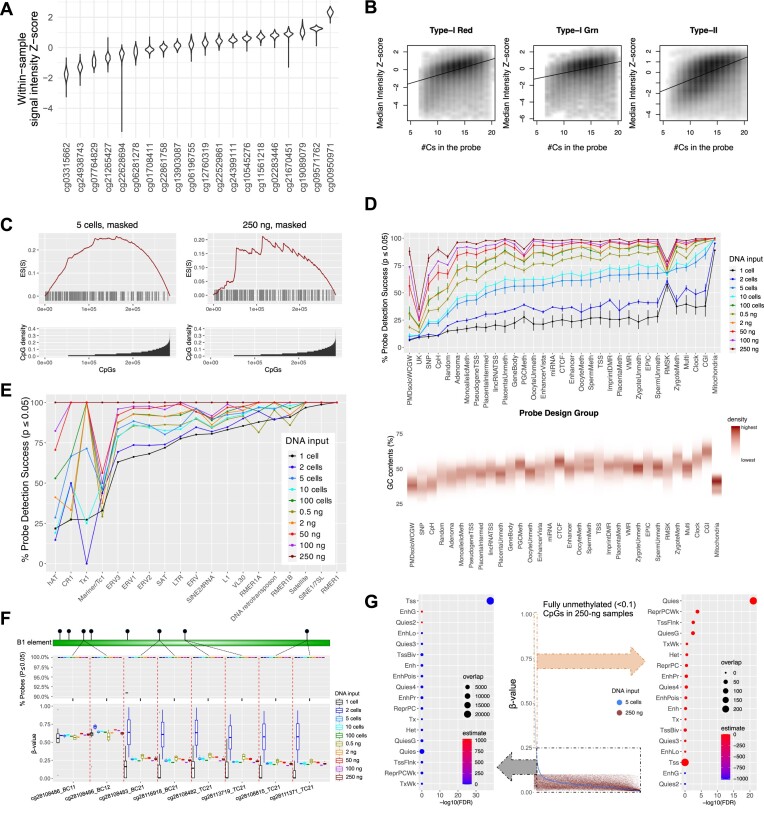
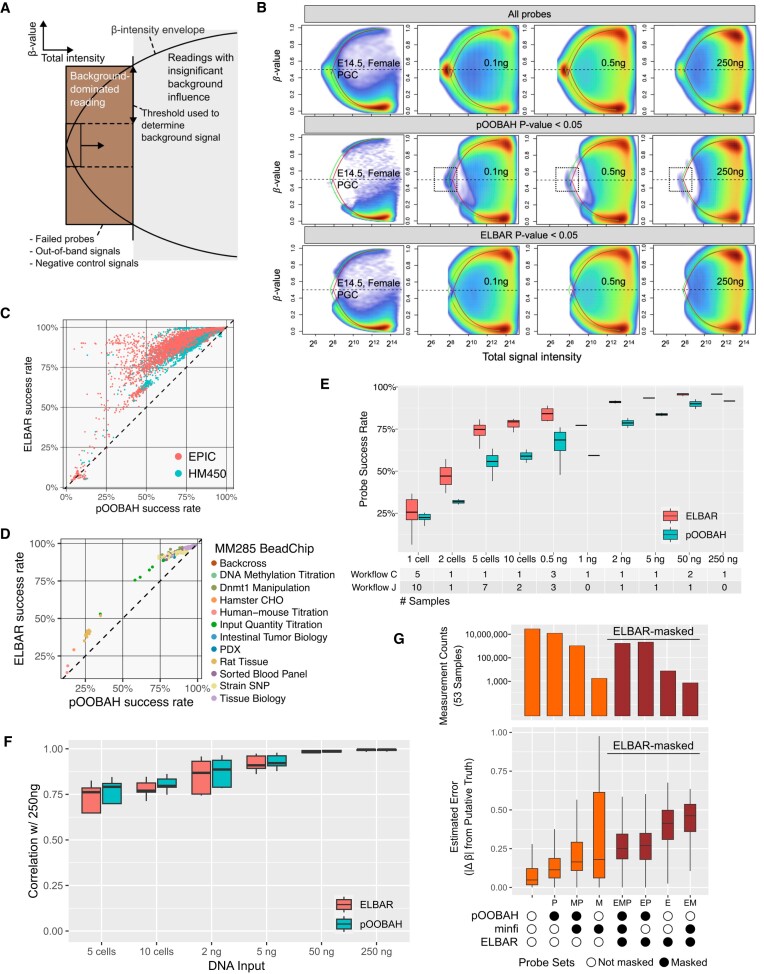
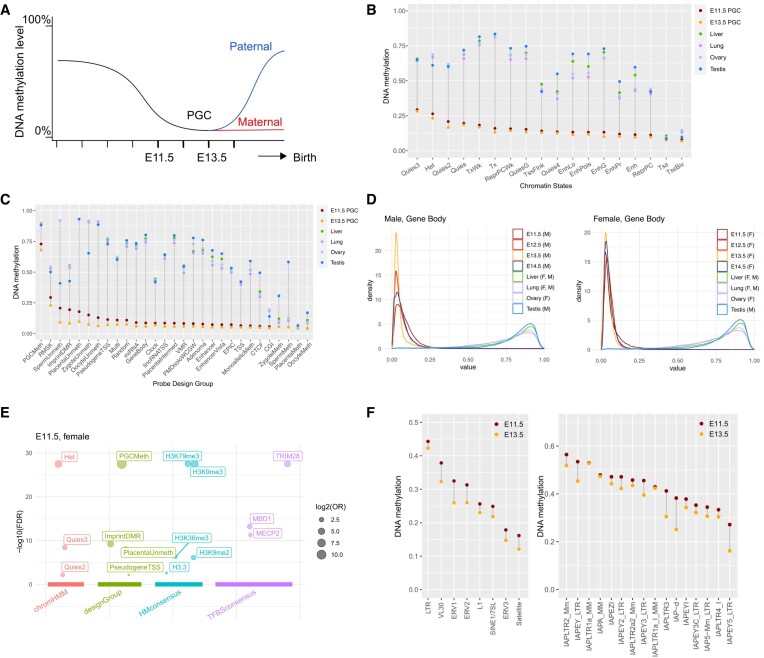
The Infinium BeadChip is the most widely used DNA methylome assay technology for population-scale epigenome profiling. However, the standard workflow requires over 200 ng of input DNA, hindering its application to small cell-number samples, such as primordial germ cells. We developed experimental and analysis workflows to extend this technology to suboptimal input DNA conditions, including ultra-low input down to single cells. DNA preamplification significantly enhanced detection rates to over 50% in five-cell samples and ∼25% in single cells. Enzymatic conversion also substantially improved data quality. Computationally, we developed a method to model the background signal's influence on the DNA methylation level readings. The modified detection P-value calculation achieved higher sensitivities for low-input datasets and was validated in over 100 000 public diverse methylome profiles. We employed the optimized workflow to query the demethylation dynamics in mouse primordial germ cells available at low cell numbers. Our data revealed nuanced chromatin states, sex disparities, and the role of DNA methylation in transposable element regulation during germ cell development. Collectively, we present comprehensive experimental and computational solutions to extend this widely used methylation assay technology to applications with limited DNA.
© The Author(s) 2024. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures
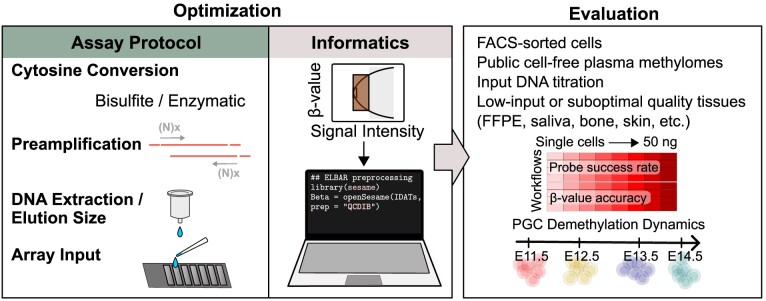
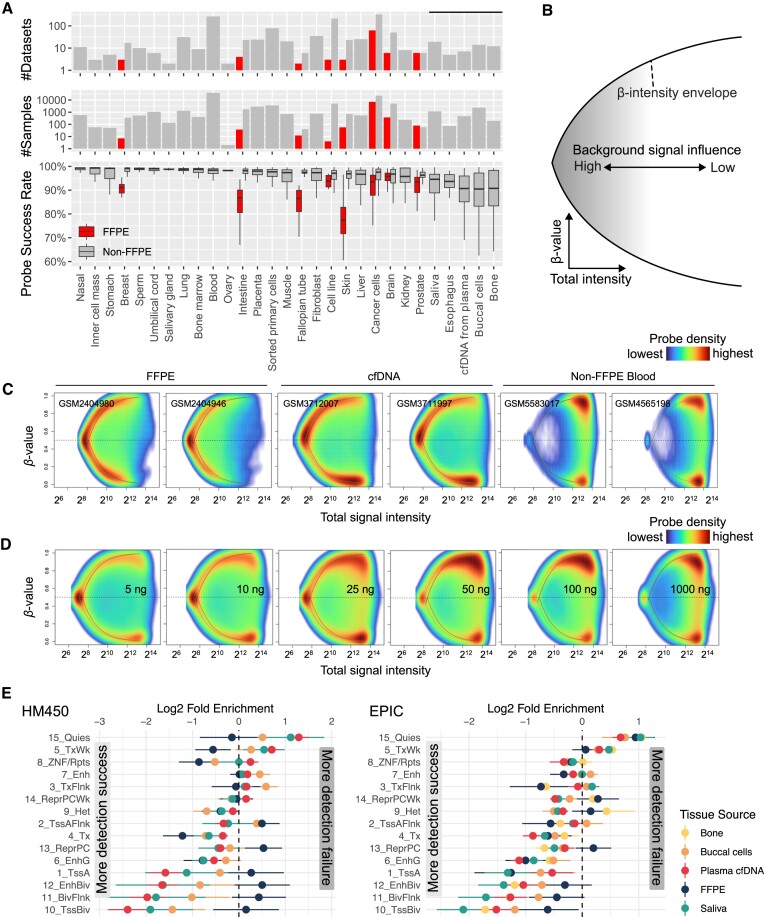
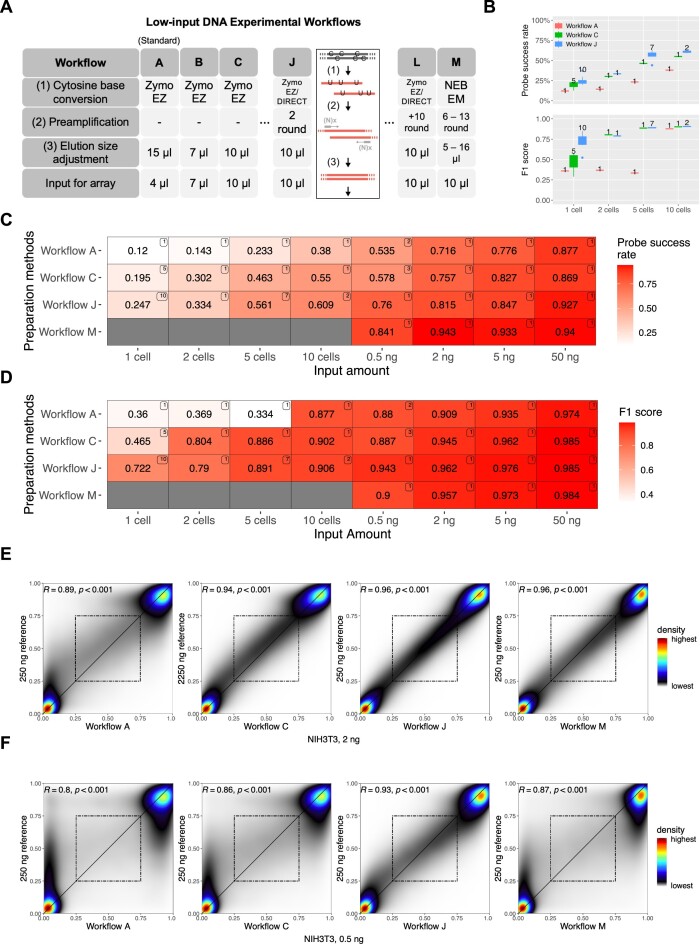
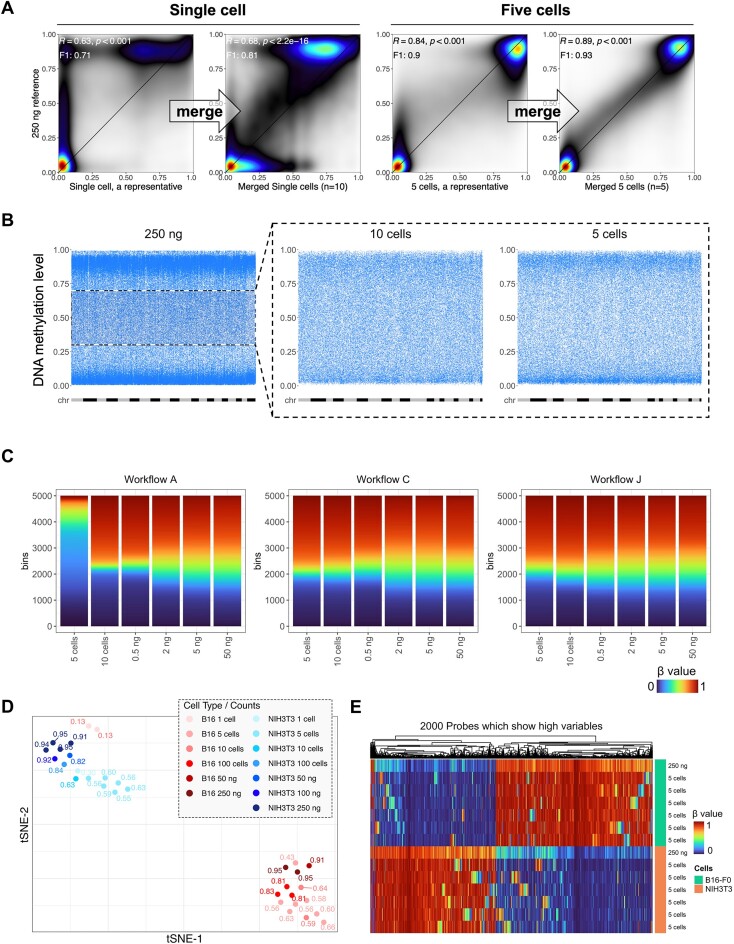
Update of
-
Low-input and single-cell methods for Infinium DNA methylation BeadChips.bioRxiv [Preprint]. 2023 Sep 22:2023.09.18.558252. doi: 10.1101/2023.09.18.558252. bioRxiv. 2023. Update in: Nucleic Acids Res. 2024 Apr 24;52(7):e38. doi: 10.1093/nar/gkae127. PMID: 37786695 Free PMC article. Updated. Preprint.
References
-
- Greenberg M.V.C., Bourc’his D The diverse roles of DNA methylation in mammalian development and disease. Nat. Rev. Mol. Cell Biol. 2019; 20:590–607. - PubMed
-
- Bibikova M., Barnes B., Tsan C., Ho V., Klotzle B., Le J.M., Delano D., Zhang L., Schroth G.P., Gunderson K.L. et al. . High density DNA methylation array with single CpG site resolution. Genomics. 2011; 98:288–295. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
