

Enhancing chimeric antigen receptor T cell therapy by modulating the p53 signaling network with Δ133p53α
- PMID: 38408246
- PMCID: PMC10927528
- DOI: 10.1073/pnas.2317735121
Enhancing chimeric antigen receptor T cell therapy by modulating the p53 signaling network with Δ133p53α
Abstract
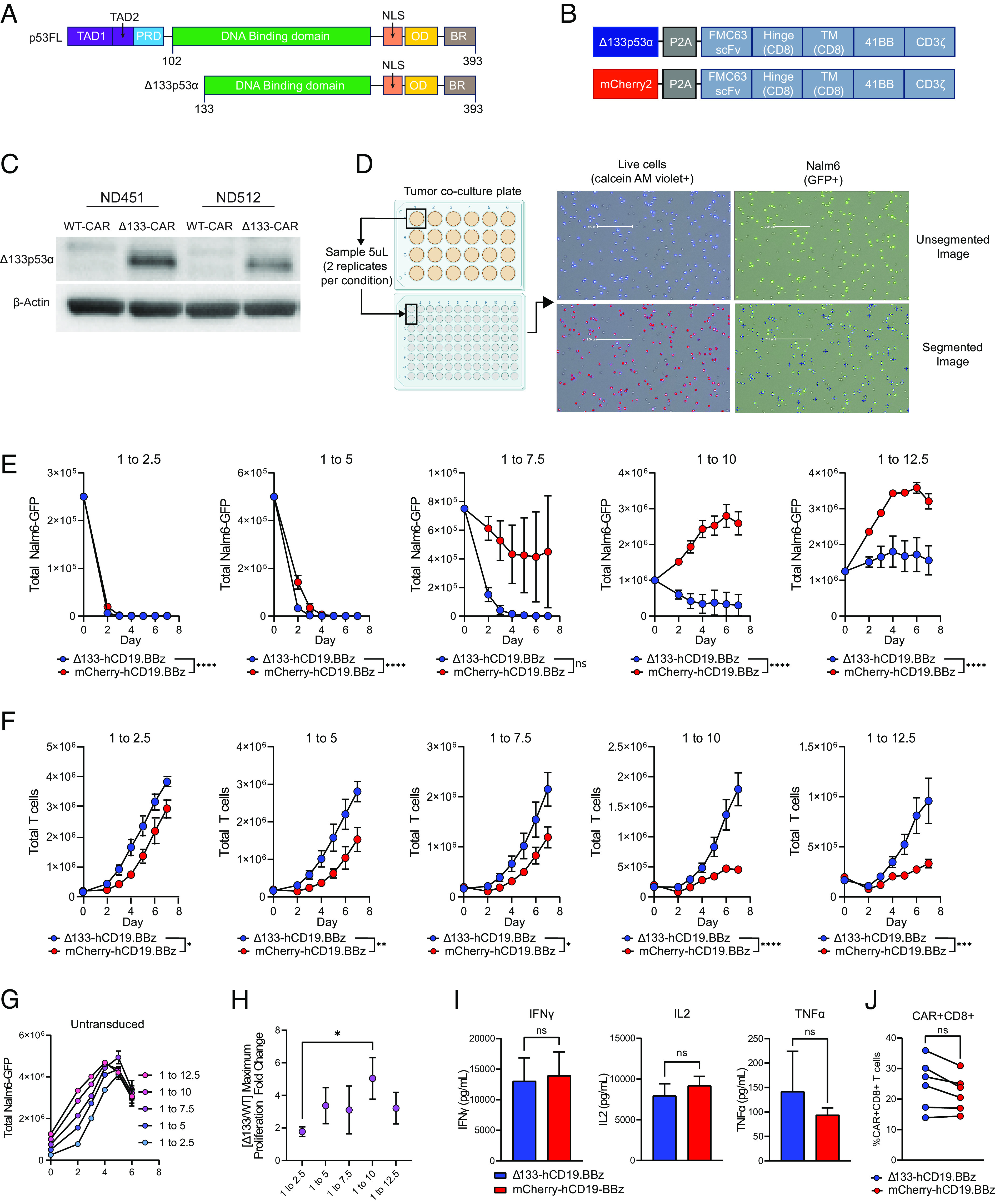
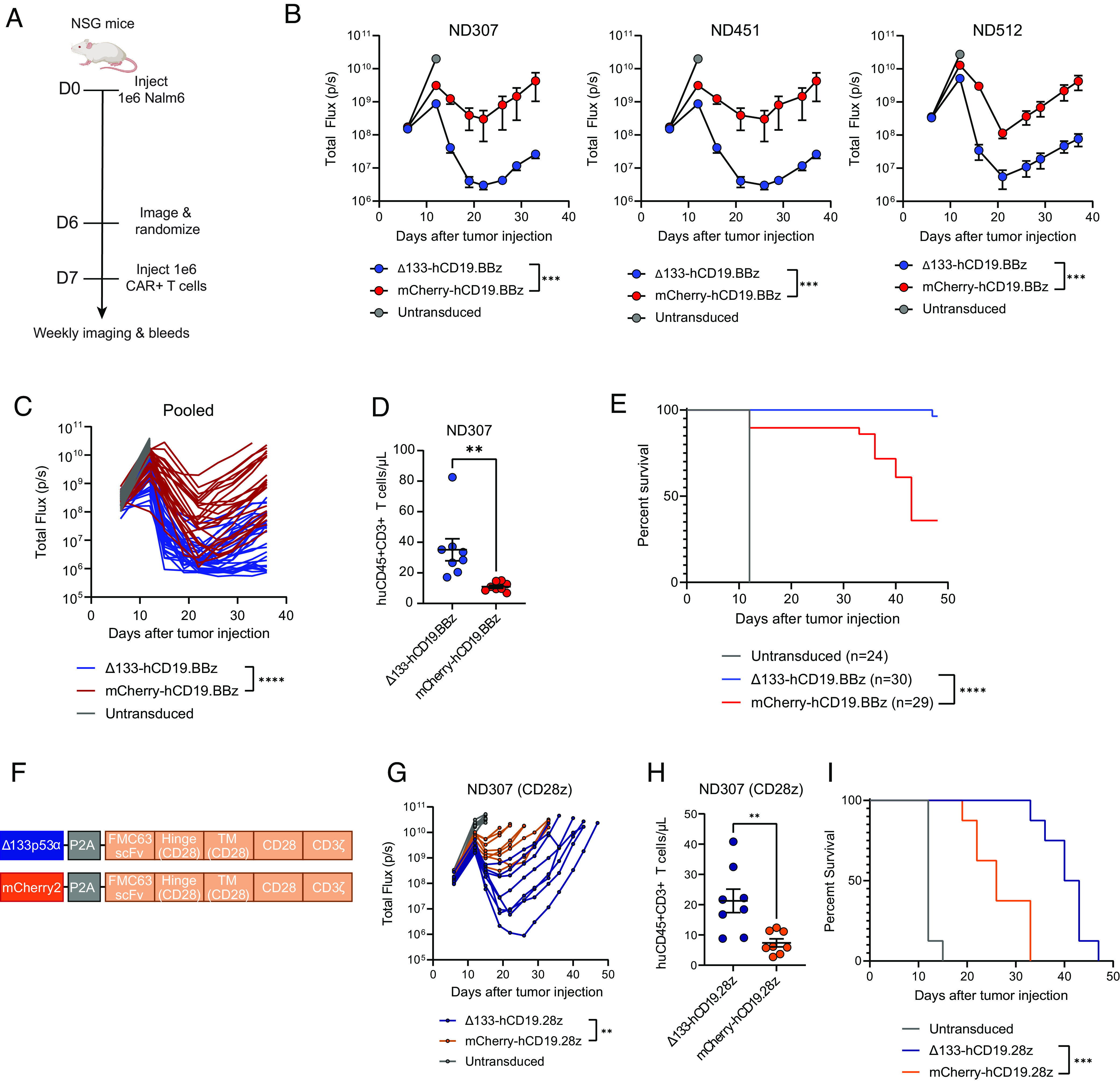
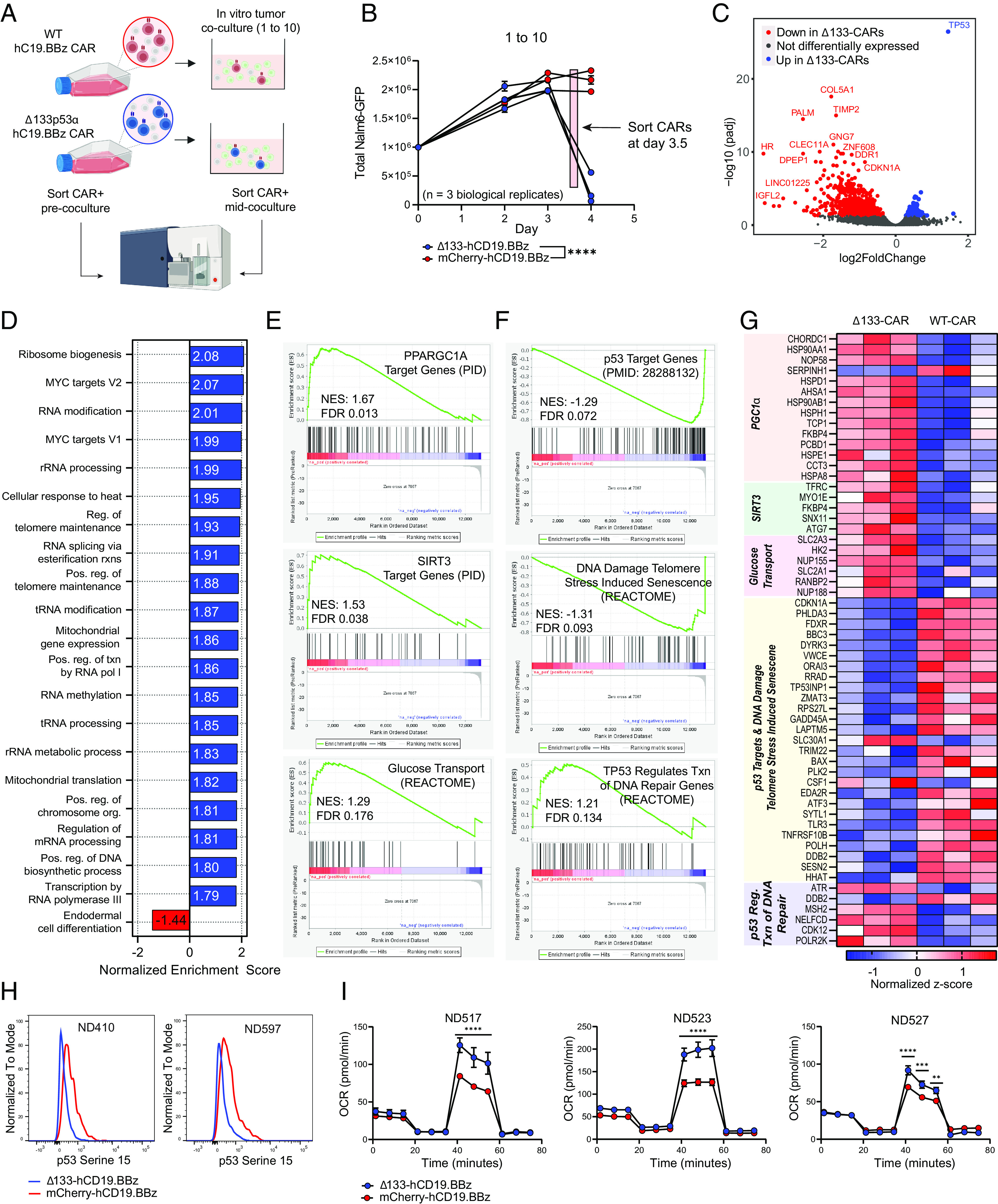
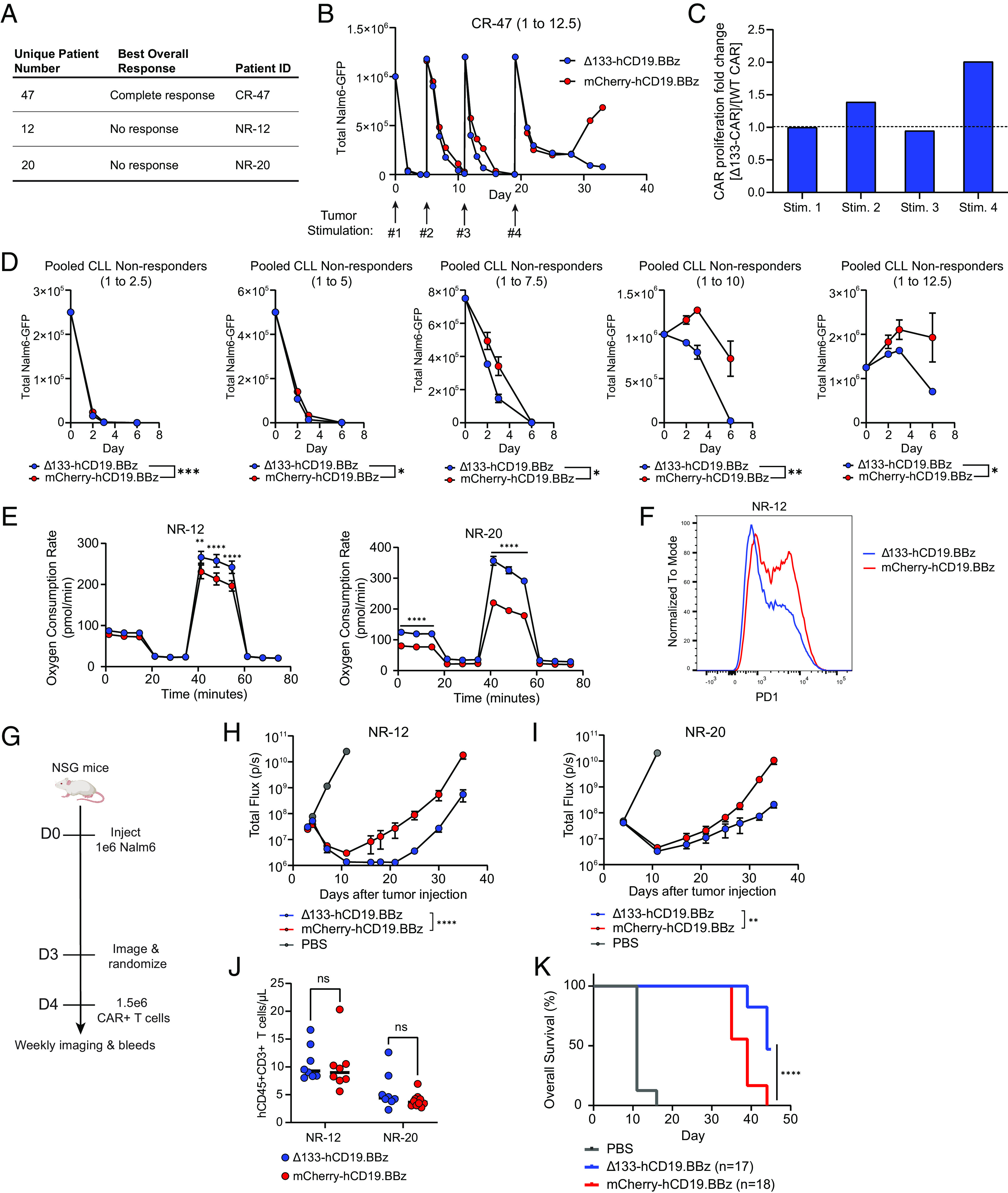
Chimeric antigen receptor (CAR) T cell dysfunction is a major barrier to achieving lasting remission in hematologic cancers, especially in chronic lymphocytic leukemia (CLL). We have shown previously that Δ133p53α, an endogenous isoform of the human TP53 gene, decreases in expression with age in human T cells, and that reconstitution of Δ133p53α in poorly functional T cells can rescue proliferation [A. M. Mondal et al., J. Clin. Invest. 123, 5247-5257 (2013)]. Although Δ133p53α lacks a transactivation domain, it can form heterooligomers with full-length p53 and modulate the p53-mediated stress response [I. Horikawa et al., Cell Death Differ. 24, 1017-1028 (2017)]. Here, we show that constitutive expression of Δ133p53α potentiates the anti-tumor activity of CD19-directed CAR T cells and limits dysfunction under conditions of high tumor burden and metabolic stress. We demonstrate that Δ133p53α-expressing CAR T cells exhibit a robust metabolic phenotype, maintaining the ability to execute effector functions and continue proliferating under nutrient-limiting conditions, in part due to upregulation of critical biosynthetic processes and improved mitochondrial function. Importantly, we show that our strategy to constitutively express Δ133p53α improves the anti-tumor efficacy of CAR T cells generated from CLL patients that previously failed CAR T cell therapy. More broadly, our results point to the potential role of the p53-mediated stress response in limiting the prolonged antitumor functions required for complete tumor clearance in patients with high disease burden, suggesting that modulation of the p53 signaling network with Δ133p53α may represent a translationally viable strategy for improving CAR T cell therapy.
Keywords: T cell; cancer therapy; senescence.
Conflict of interest statement
Competing interests statement:C.H.J. is an inventor of patents related to the CAR therapy product which is the subject of this paper, as well as other CAR therapy products, and may be eligible to receive a select portion of royalties paid from Kite to the University of Pennsylvania. C.H.J. is a scientific co-founder and holds equity in Capstan Therapeutics, Dispatch Biotherapeutics and Bluewhale Bio. C.H.J. serves on the board of AC Immune and is a scientific advisor to BluesphereBio, Cabaletta, Carisma, Cartography, Cellares, Cellcarta, Celldex, Danaher, Decheng, ImmuneSensor, Kite, Marble Therapeutics, Poseida, Verismo, Viracta, and WIRB-Copernicus group. C.H.J., R.M.Y., and J.S. are inventors on patents and/or patent applications licensed to Novartis Institutes of Biomedical Research and Kite and may receive license revenue from such licenses. R.O.C. is an inventor on patents licensed to Novartis of Biomedical Research and has equity in Nucleus Biologics and Stoic Bio. R.O.C. is also a scientific advisor to Nucleus Biologics. C.R. consults for Marble Therapeutics and is a scientific advisor to Boston Labs.
Figures
Comment in
-
Improving T cell killing and understanding senescence: Possible roles for TP53 in cancer immunotherapy.Proc Natl Acad Sci U S A. 2024 Mar 19;121(12):e2402533121. doi: 10.1073/pnas.2402533121. Epub 2024 Mar 11. Proc Natl Acad Sci U S A. 2024. PMID: 38466858 Free PMC article. No abstract available.
Similar articles
-
In Vivo Expansion and Antitumor Activity of Coinfused CD28- and 4-1BB-Engineered CAR-T Cells in Patients with B Cell Leukemia.Mol Ther. 2018 Apr 4;26(4):976-985. doi: 10.1016/j.ymthe.2018.01.022. Epub 2018 Feb 2. Mol Ther. 2018. PMID: 29503204 Free PMC article. Clinical Trial.
-
Improving therapy of chronic lymphocytic leukemia with chimeric antigen receptor T cells.Semin Oncol. 2016 Apr;43(2):291-9. doi: 10.1053/j.seminoncol.2016.02.006. Epub 2016 Feb 9. Semin Oncol. 2016. PMID: 27040708 Free PMC article. Review.
-
Δ133p53α enhances metabolic and cellular fitness of TCR-engineered T cells and promotes superior antitumor immunity.J Immunother Cancer. 2021 Jun;9(6):e001846. doi: 10.1136/jitc-2020-001846. J Immunother Cancer. 2021. PMID: 34112738 Free PMC article.
-
PiggyBac Transposon-Mediated CD19 Chimeric Antigen Receptor-T Cells Derived From CD45RA-Positive Peripheral Blood Mononuclear Cells Possess Potent and Sustained Antileukemic Function.Front Immunol. 2022 Jan 27;13:770132. doi: 10.3389/fimmu.2022.770132. eCollection 2022. Front Immunol. 2022. PMID: 35154098 Free PMC article.
-
CAR T Cell Therapy in Acute Lymphoblastic Leukemia and Potential for Chronic Lymphocytic Leukemia.Curr Treat Options Oncol. 2016 Jun;17(6):28. doi: 10.1007/s11864-016-0406-4. Curr Treat Options Oncol. 2016. PMID: 27098534 Review.
Cited by
-
Improving T cell killing and understanding senescence: Possible roles for TP53 in cancer immunotherapy.Proc Natl Acad Sci U S A. 2024 Mar 19;121(12):e2402533121. doi: 10.1073/pnas.2402533121. Epub 2024 Mar 11. Proc Natl Acad Sci U S A. 2024. PMID: 38466858 Free PMC article. No abstract available.
-
Δ133p53α-mediated inhibition of astrocyte senescence and neurotoxicity as a possible therapeutic approach for neurodegenerative diseases.Neuroscience. 2025 Aug 6;580:54-61. doi: 10.1016/j.neuroscience.2025.06.031. Epub 2025 Jun 14. Neuroscience. 2025. PMID: 40523602 Free PMC article. Review.
-
Impact of mitochondrial metabolism on T-cell dysfunction in chronic lymphocytic leukemia.Front Cell Dev Biol. 2025 Apr 17;13:1577081. doi: 10.3389/fcell.2025.1577081. eCollection 2025. Front Cell Dev Biol. 2025. PMID: 40313718 Free PMC article. Review.
-
The senescence-inhibitory p53 isoform Δ133p53α: enhancing cancer immunotherapy and exploring novel therapeutic approaches for senescence-associated diseases.Geroscience. 2025 Aug 6. doi: 10.1007/s11357-025-01819-y. Online ahead of print. Geroscience. 2025. PMID: 40770529 Review.
-
Mitigating T-cell mitochondrial dysfunction in CLL to augment CAR T-cell therapy: evaluation in an immunocompetent model.Blood Adv. 2025 May 27;9(10):2511-2529. doi: 10.1182/bloodadvances.2024014822. Blood Adv. 2025. PMID: 39938006 Free PMC article.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
