

Interplay between coding and non-coding regulation drives the Arabidopsis seed-to-seedling transition
- PMID: 38409232
- PMCID: PMC10897432
- DOI: 10.1038/s41467-024-46082-5
Interplay between coding and non-coding regulation drives the Arabidopsis seed-to-seedling transition
Abstract
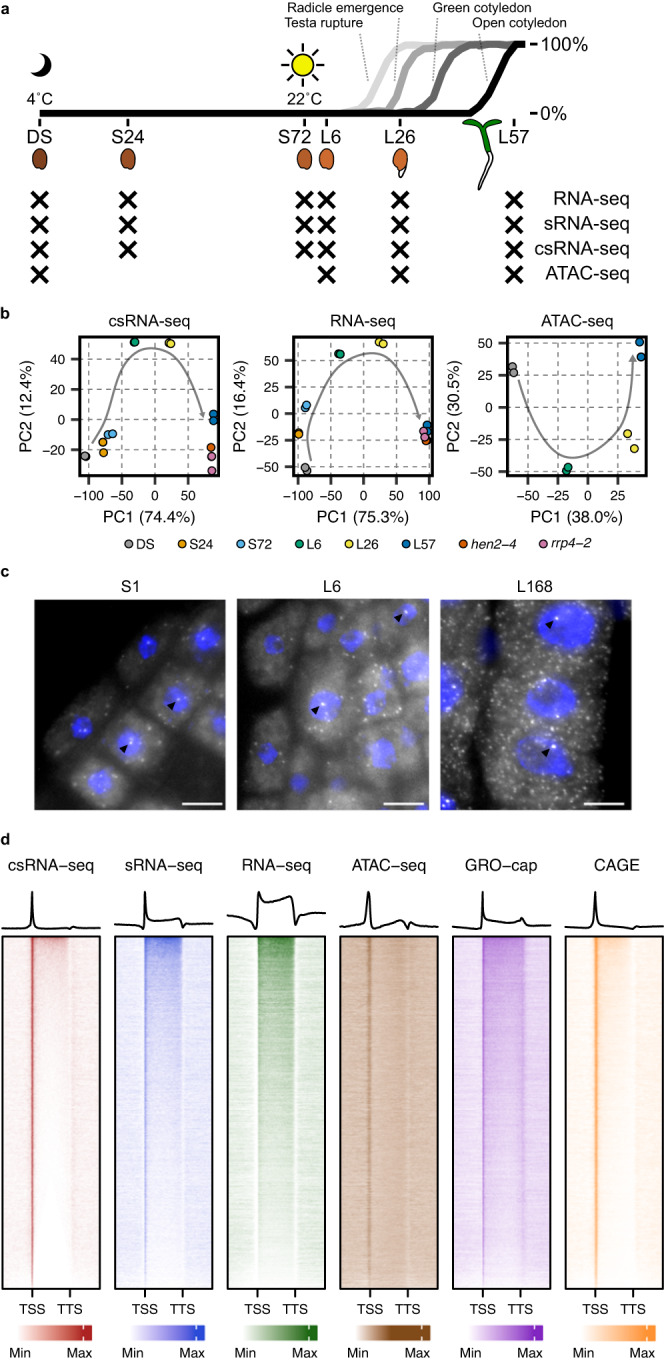
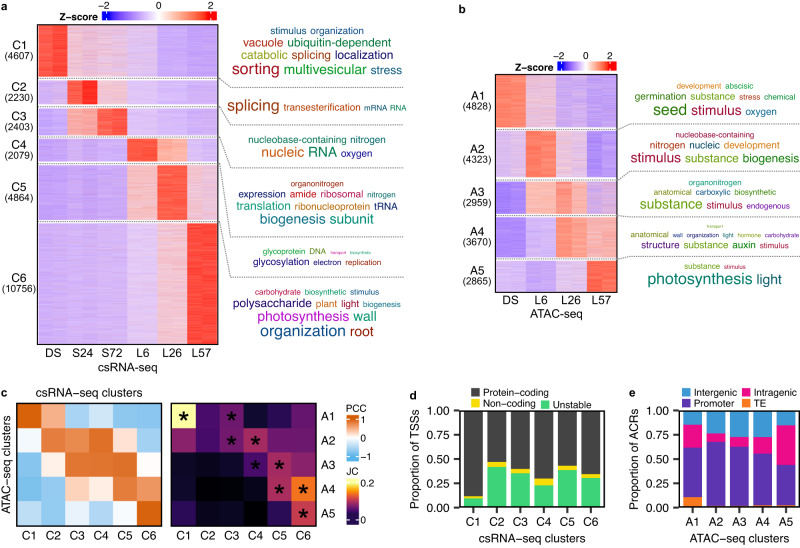
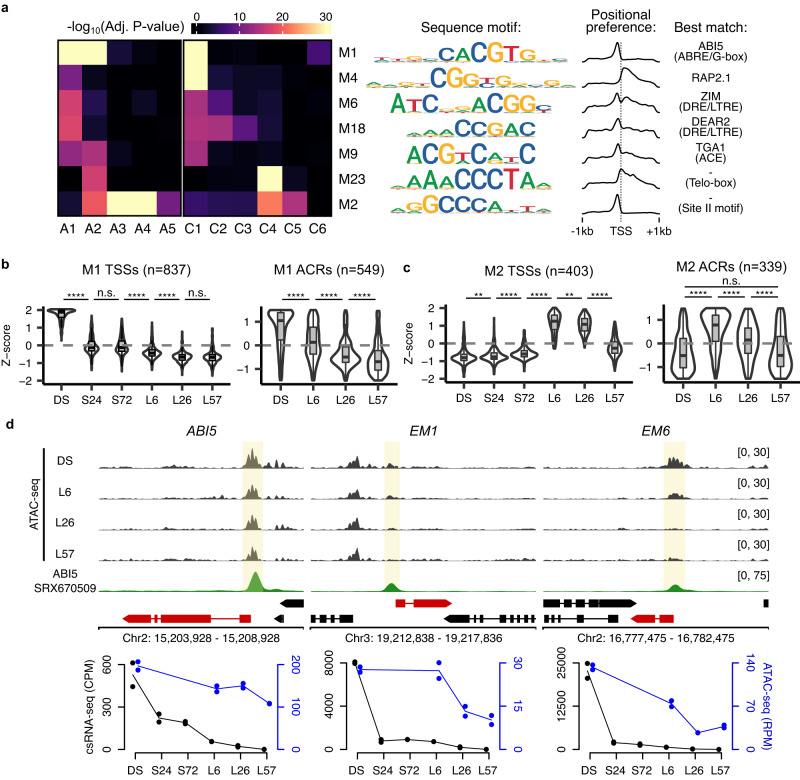
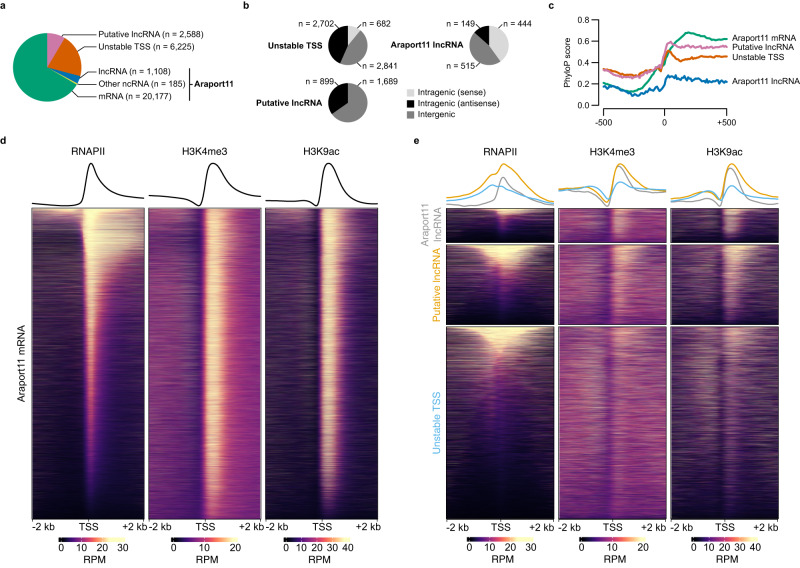
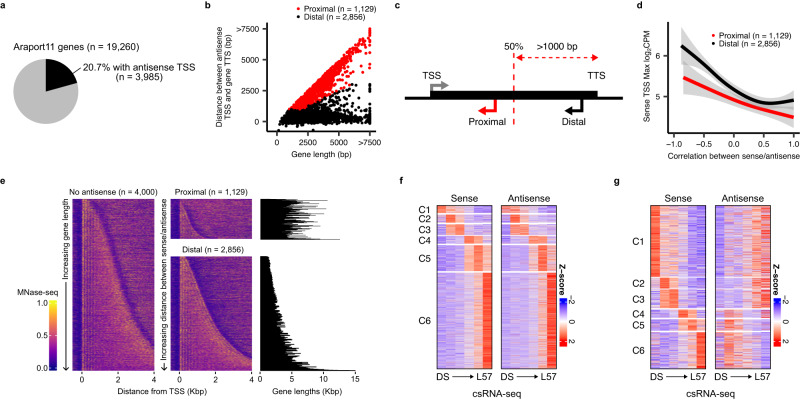
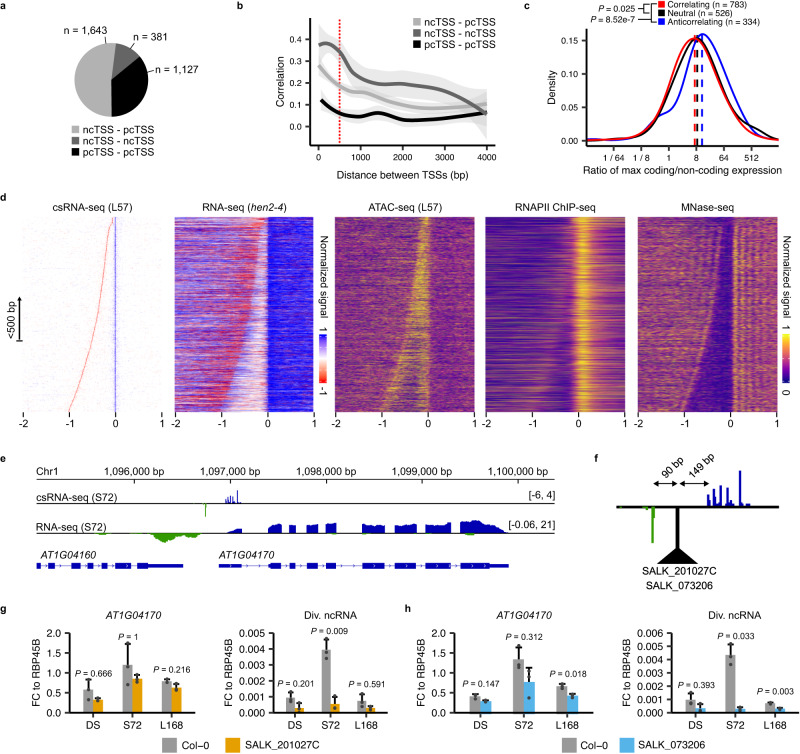
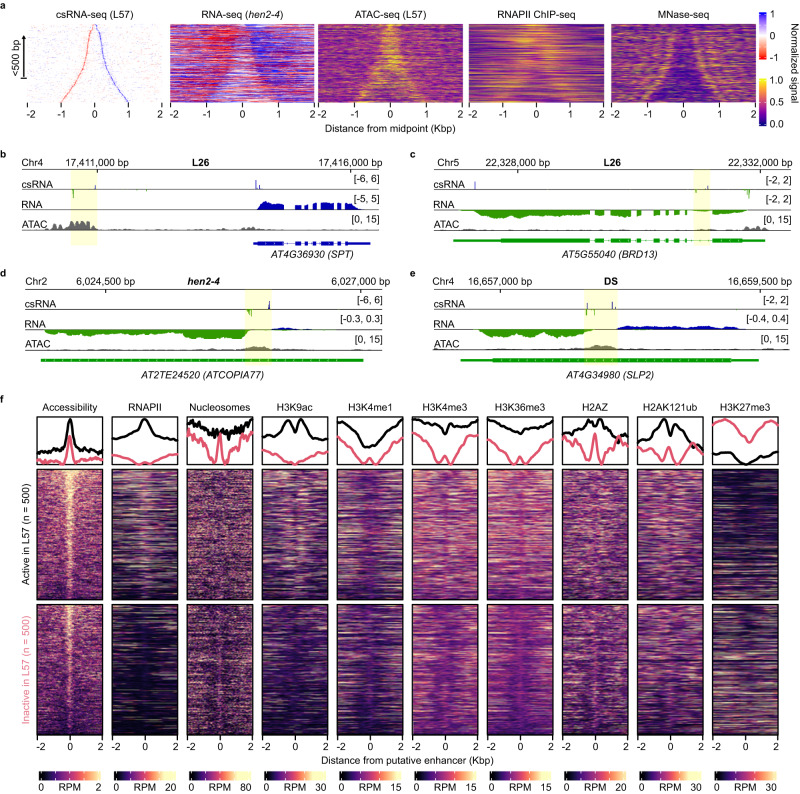
Translation of seed stored mRNAs is essential to trigger germination. However, when RNAPII re-engages RNA synthesis during the seed-to-seedling transition has remained in question. Combining csRNA-seq, ATAC-seq and smFISH in Arabidopsis thaliana we demonstrate that active transcription initiation is detectable during the entire germination process. Features of non-coding regulation such as dynamic changes in chromatin accessible regions, antisense transcription, as well as bidirectional non-coding promoters are widespread throughout the Arabidopsis genome. We show that sensitivity to exogenous ABSCISIC ACID (ABA) during germination depends on proximal promoter accessibility at ABA-responsive genes. Moreover, we provide genetic validation of the existence of divergent transcription in plants. Our results reveal that active enhancer elements are transcribed producing non-coding enhancer RNAs (eRNAs) as widely documented in metazoans. In sum, this study defining the extent and role of coding and non-coding transcription during key stages of germination expands our understanding of transcriptional mechanisms underlying plant developmental transitions.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Nakabayashi K, Okamoto M, Koshiba T, Kamiya Y, Nambara E. Genome-wide profiling of stored mRNA in Arabidopsis thaliana seed germination: Epigenetic and genetic regulation of transcription in seed. Plant J. 2005;41:697–709. - PubMed
MeSH terms
Substances
Grants and funding
- LCF/BQ/ PI19/11690003/"la Caixa" Foundation (Caixa Foundation)
- PRE2019-090156/Ministry of Economy and Competitiveness | Agencia Estatal de Investigación (Spanish Agencia Estatal de Investigación)
- SEV-2015-0533/Ministry of Economy and Competitiveness | Agencia Estatal de Investigación (Spanish Agencia Estatal de Investigación)
- EUR2021-122003/Ministry of Economy and Competitiveness | Agencia Estatal de Investigación (Spanish Agencia Estatal de Investigación)
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
