

E-cadherin loss drives diffuse-type gastric tumorigenesis via EZH2-mediated reprogramming
- PMID: 38411616
- PMCID: PMC10899090
- DOI: 10.1084/jem.20230561
E-cadherin loss drives diffuse-type gastric tumorigenesis via EZH2-mediated reprogramming
Abstract
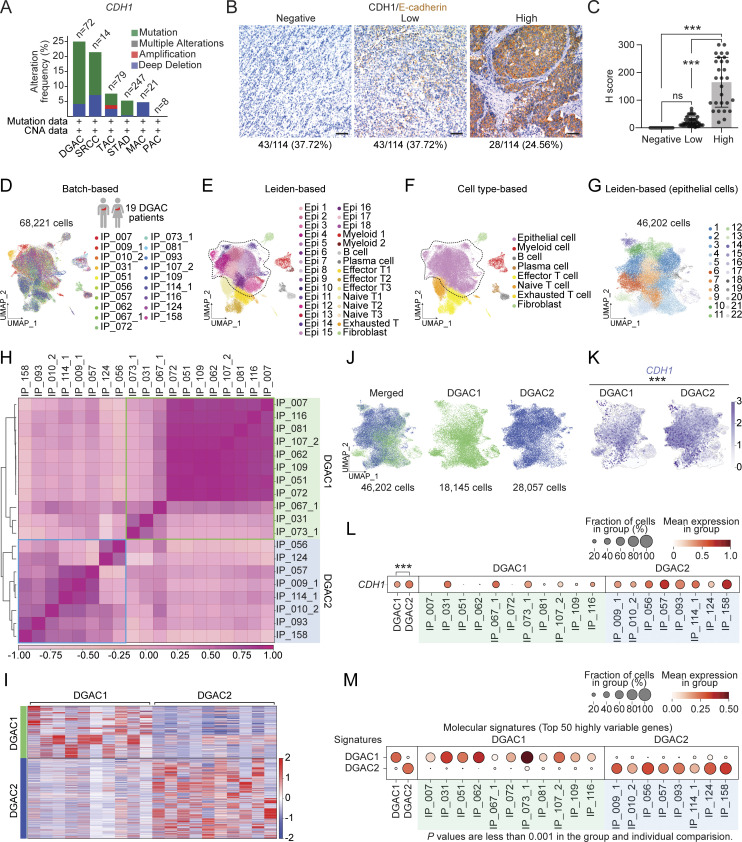
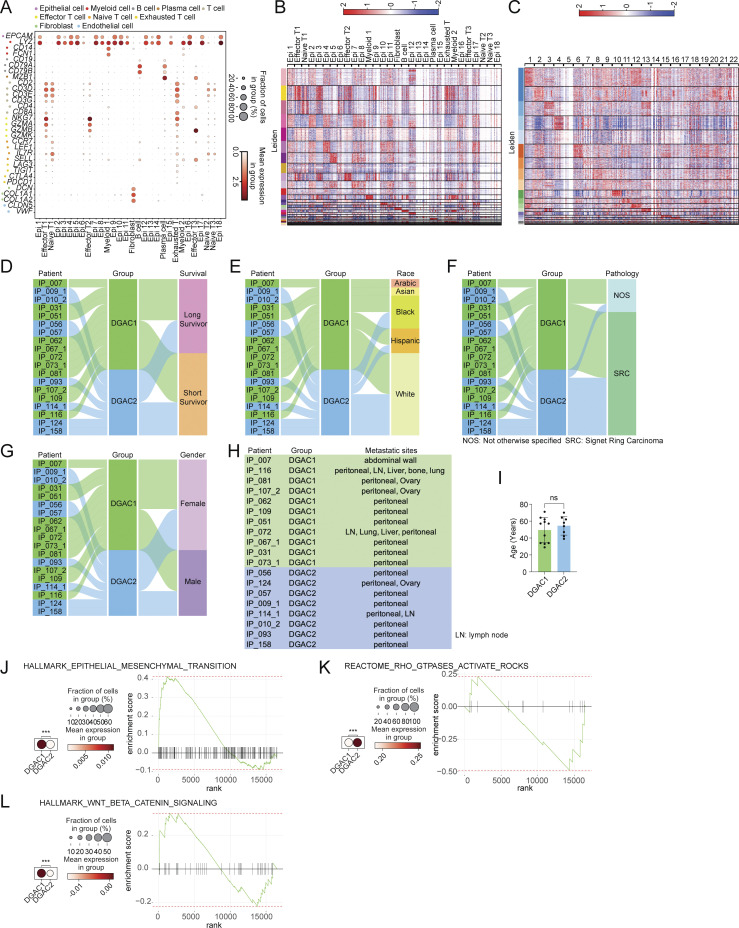
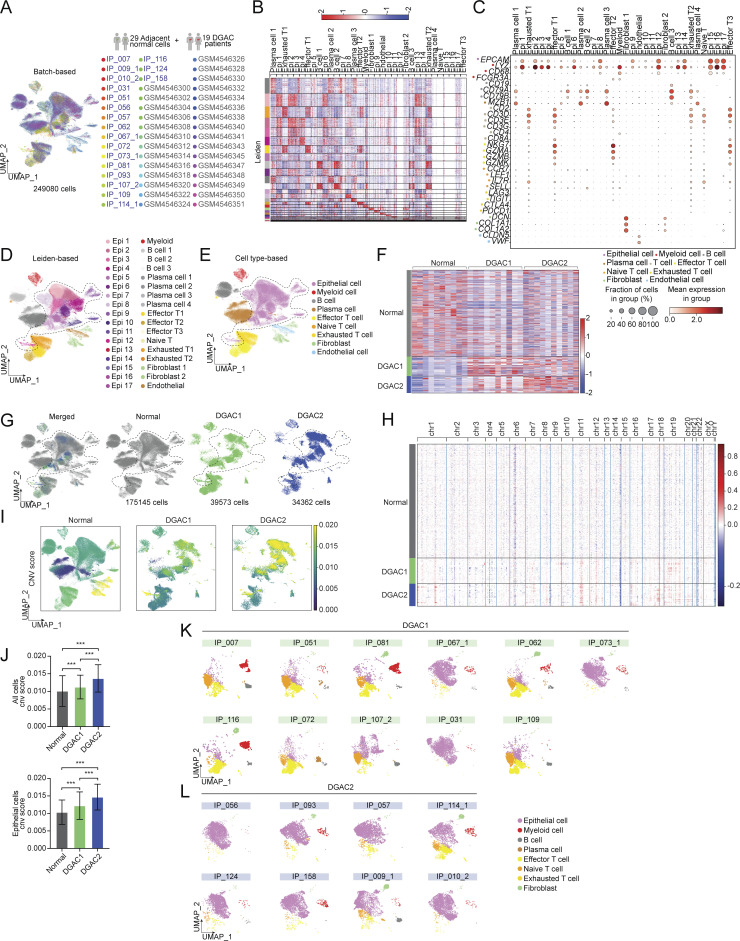
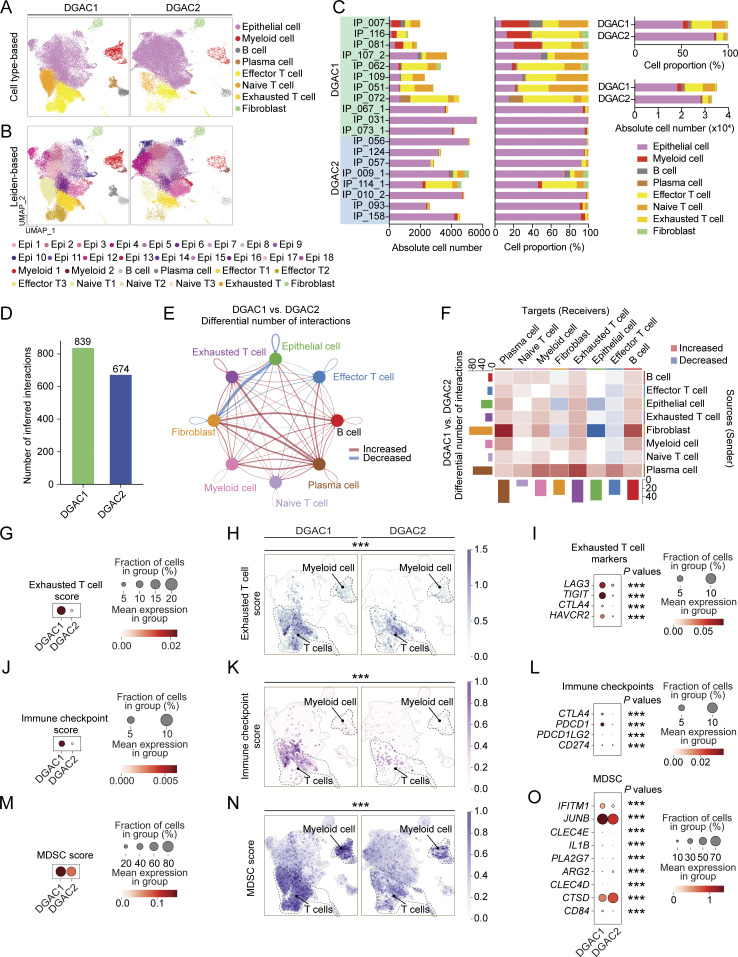
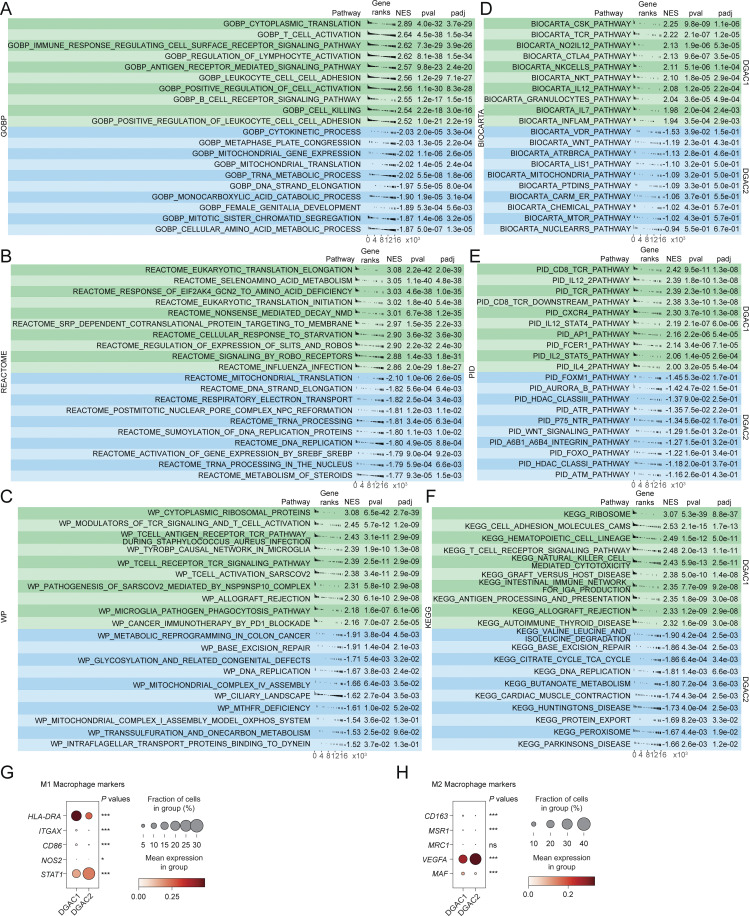
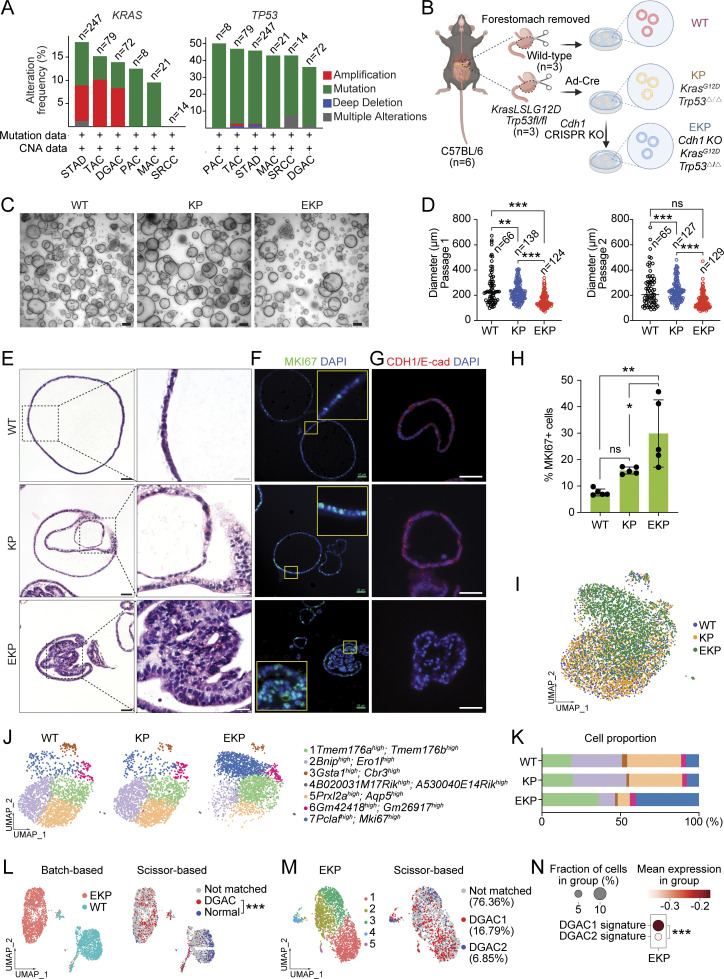
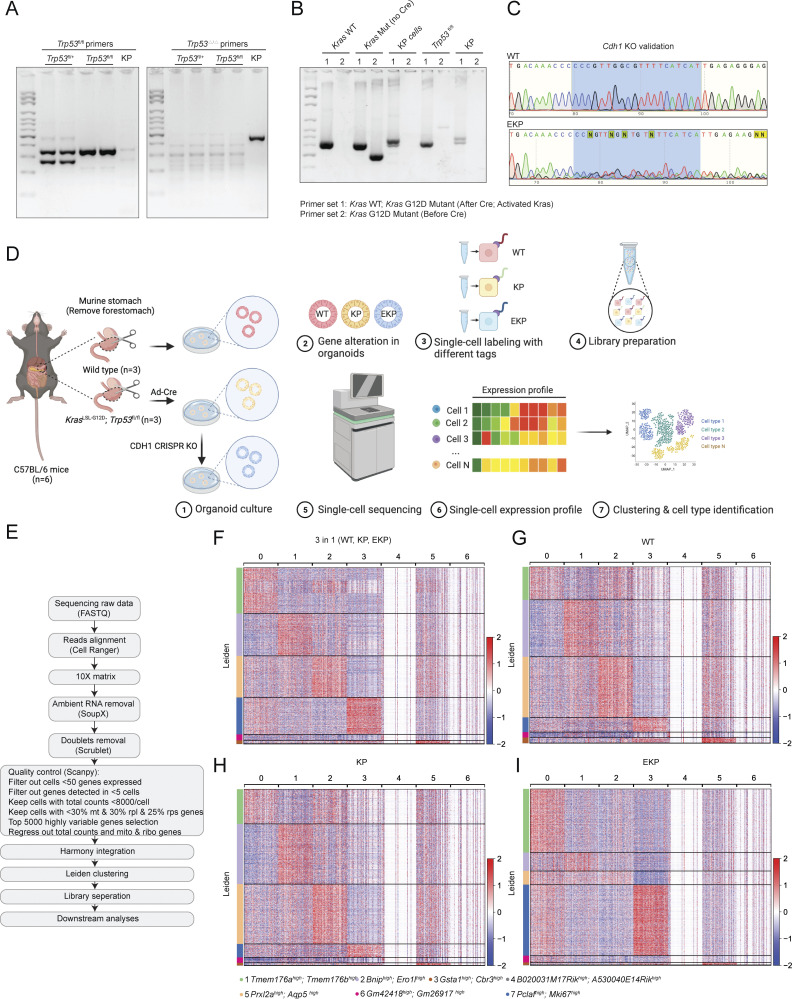
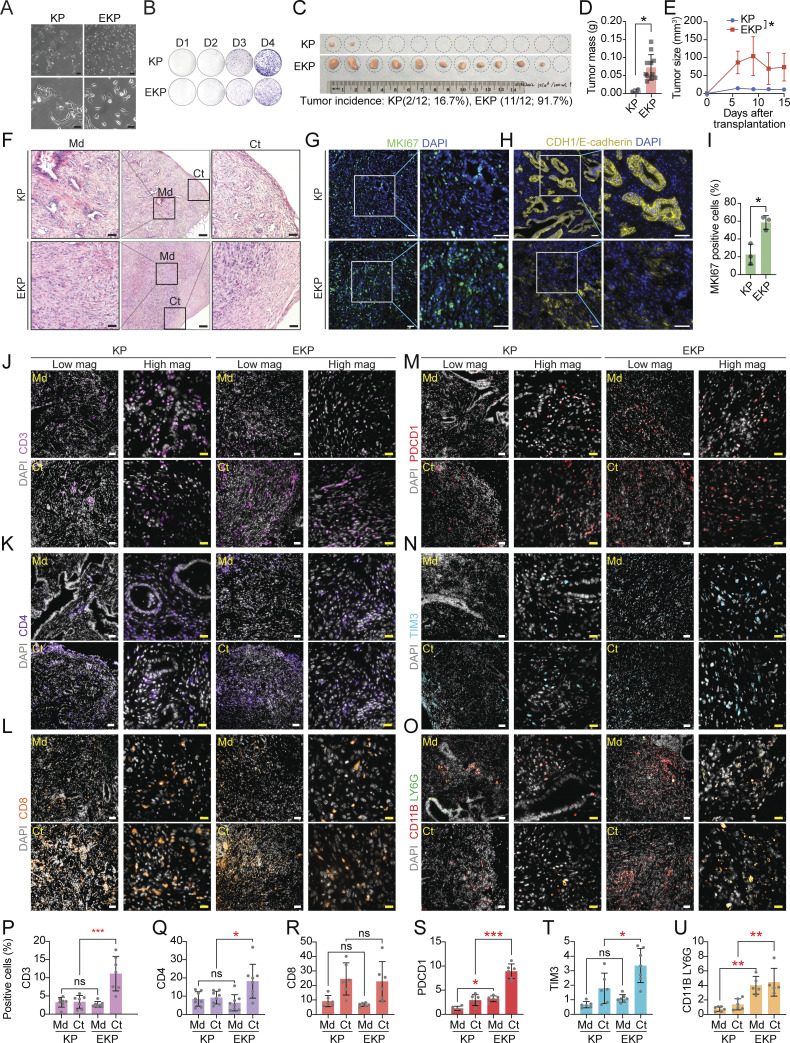
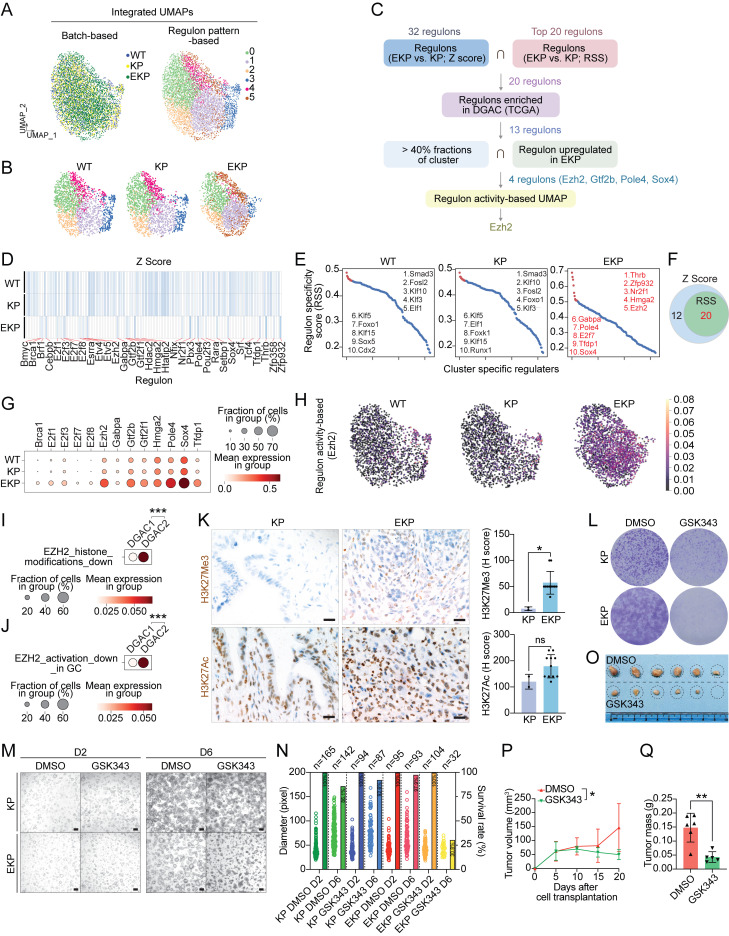
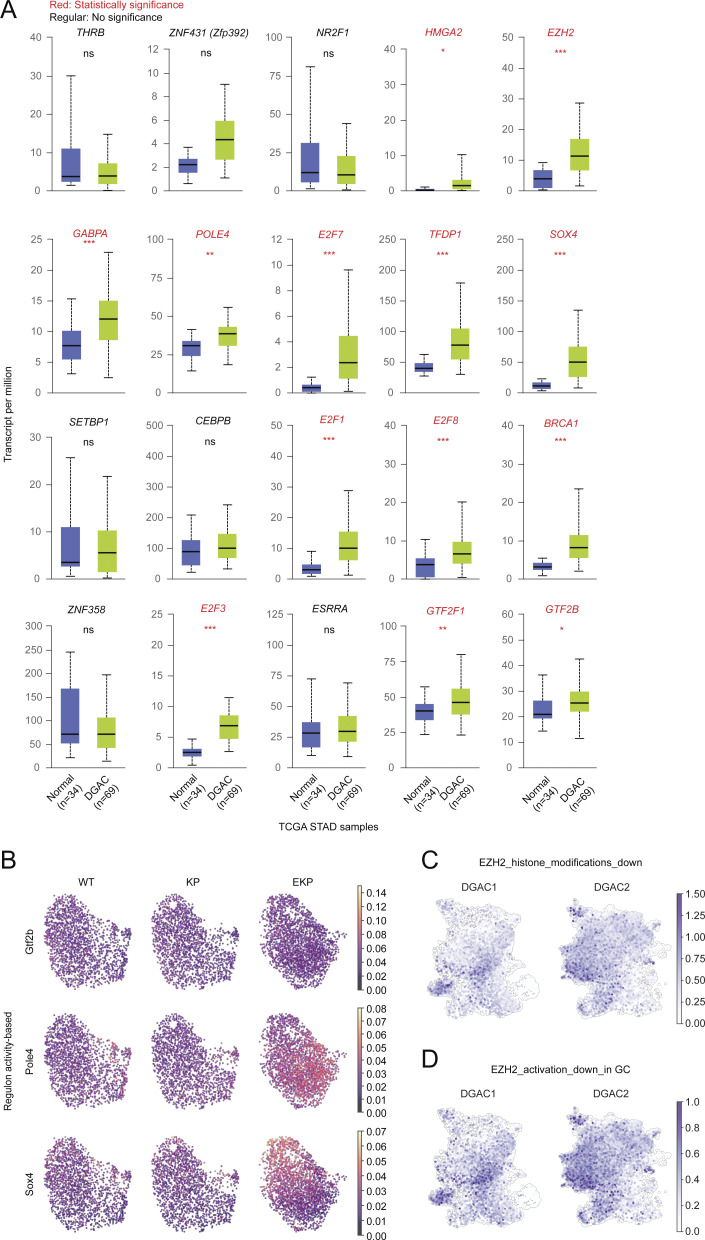
Diffuse-type gastric adenocarcinoma (DGAC) is a deadly cancer often diagnosed late and resistant to treatment. While hereditary DGAC is linked to CDH1 mutations, the role of CDH1/E-cadherin inactivation in sporadic DGAC tumorigenesis remains elusive. We discovered CDH1 inactivation in a subset of DGAC patient tumors. Analyzing single-cell transcriptomes in malignant ascites, we identified two DGAC subtypes: DGAC1 (CDH1 loss) and DGAC2 (lacking immune response). DGAC1 displayed distinct molecular signatures, activated DGAC-related pathways, and an abundance of exhausted T cells in ascites. Genetically engineered murine gastric organoids showed that Cdh1 knock-out (KO), KrasG12D, Trp53 KO (EKP) accelerates tumorigenesis with immune evasion compared with KrasG12D, Trp53 KO (KP). We also identified EZH2 as a key mediator promoting CDH1 loss-associated DGAC tumorigenesis. These findings highlight DGAC's molecular diversity and potential for personalized treatment in CDH1-inactivated patients.
© 2024 Zou et al.
Conflict of interest statement
Disclosures: The authors declare no competing interests exist.
Figures
Update of
-
CDH1 loss promotes diffuse-type gastric cancer tumorigenesis via epigenetic reprogramming and immune evasion.bioRxiv [Preprint]. 2023 Oct 11:2023.03.23.533976. doi: 10.1101/2023.03.23.533976. bioRxiv. 2023. Update in: J Exp Med. 2024 Apr 1;221(4):e20230561. doi: 10.1084/jem.20230561. PMID: 36993615 Free PMC article. Updated. Preprint.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
