

Framework for in vivo T cell screens
- PMID: 38411617
- PMCID: PMC10899089
- DOI: 10.1084/jem.20230699
Framework for in vivo T cell screens
Abstract
In vivo T cell screens are a powerful tool for elucidating complex mechanisms of immunity, yet there is a lack of consensus on the screen design parameters required for robust in vivo screens: gene library size, cell transfer quantity, and number of mice. Here, we describe the Framework for In vivo T cell Screens (FITS) to provide experimental and analytical guidelines to determine optimal parameters for diverse in vivo contexts. As a proof-of-concept, we used FITS to optimize the parameters for a CD8+ T cell screen in the B16-OVA tumor model. We also included unique molecular identifiers (UMIs) in our screens to (1) improve statistical power and (2) track T cell clonal dynamics for distinct gene knockouts (KOs) across multiple tissues. These findings provide an experimental and analytical framework for performing in vivo screens in immune cells and illustrate a case study for in vivo T cell screens with UMIs.
© 2024 Milling et al.
Conflict of interest statement
Disclosures: L.E. Milling and M.W. LaFleur reported grants from Merck Sharp & Dohme LLC during the conduct of the study. N.M. Derosia and P. Prathima reported grants from Merck during the conduct of the study and grants from Merck outside the submitted work. I.S.L. Streeter and A.M. Lemmen reported grants from Merck during the conduct of the study. G.H. Hickok reported grants from Merck & Co. during the conduct of the study. N. Hacohen reported personal fees from Danger Bio/Related Science, Immune Repertoire Medicines, and CytoReason, grants from Bristol Myers Squibb, and grants from Calico Life Sciences outside the submitted work. J.G. Doench reported personal fees from BioNTech, Tango Therapeutics, Microsoft Research, and Pfizer outside the submitted work; in addition, J.G. Doench consults for Microsoft Research, Abata Therapeutics, Servier, Maze Therapeutics, BioNTech, Sangamo, and Pfizer; consults for and has equity in Tango Therapeutics; serves as a paid scientific advisor to the Laboratory for Genomics Research, funded in part by GlaxoSmithKline; and receives funding support from the Functional Genomics Consortium: Abbvie, Bristol Myers Squibb, Janssen, Vir Biotechnology, and Merck Sharp & Dohme LLC, a subsidiary of Merck & Co., Inc., Rahway, NJ, USA. J.G. Doench’s interests were reviewed and managed by the Broad Institute in accordance with its conflict-of-interest policies. A.H. Sharpe reported grants from NIH U19AI133524 and grants from NIH P01 AI108545 during the conduct of the study; grants from Vertex, Moderna, Merck Sharp & Dohme, AbbVie, Quark/IOME, Roche, Ipsen, Novartis, Erasca, Taiwan Bio, and Calico; personal fees from Surface Oncology, Sqz Biotechnologies, Elpiscience, Selecta, Bicara, Fibrogen, Alixia, GlaxoSmithKline, Janssen, Amgen, and Bioentre; and “other” from IOME, Monopteros, and Corner Therapeutics outside the submitted work; in addition, A.H. Sharpe had patent numbers 7,432,059 and 7,722,868 with royalties paid “Roche, Merck, Bristol-Myers-Squibb, EMD-Serono, Boehringer-Ingelheim, AstraZeneca, Leica, Mayo Clinic, Dako and Novartis,” patent numbers 8,652,465 and 9,457,080 licensed “Roche,” patent numbers 9,683,048, 9,815,898, 9,845,356, 10,202,454, and 10,457,733 licensed “Novartis,” patent numbers 9,580,684, 9,988,452, 10,370,446, 10,457,733, 10,752,687, 10,851,165, 10,934,353, and 15,314,251 issued; and is on scientific advisory boards for the Massachusetts General Cancer Center, Program in Cellular and Molecular Medicine at Boston Children’s Hospital, the Human Oncology and Pathogenesis Program at Memorial Sloan Kettering Cancer Center, The Gladstone Institute, and the Bloomberg-Kimmel Institute for Cancer Immunotherapy is an academic editor for the
Figures


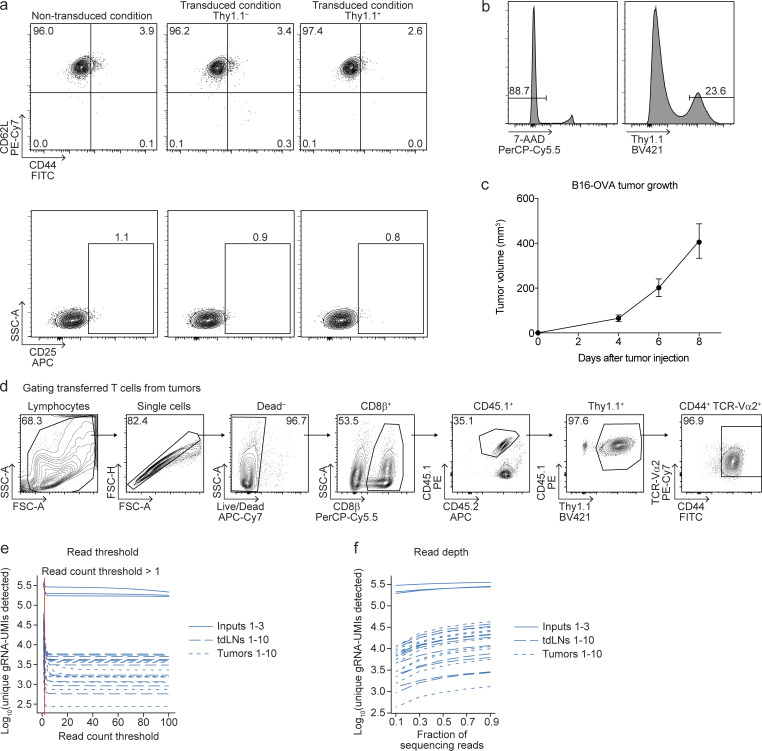
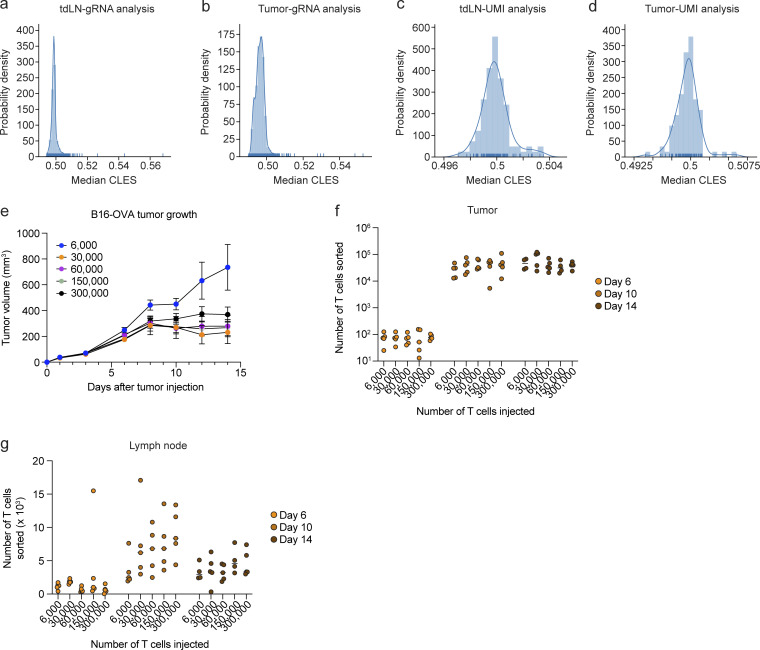








References
-
- Connolly, K.A., Kuchroo M., Venkat A., Khatun A., Wang J., William I., Hornick N.I., Fitzgerald B.L., Damo M., Kasmani M.Y., et al. 2021. A reservoir of stem-like CD8+ T cells in the tumor-draining lymph node preserves the ongoing antitumor immune response. Sci. Immunol. 6:eabg7836. 10.1126/sciimmunol.abg7836 - DOI - PMC - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
