

Nutrients: Signal 4 in T cell immunity
- PMID: 38411744
- PMCID: PMC10899091
- DOI: 10.1084/jem.20221839
Nutrients: Signal 4 in T cell immunity
Abstract
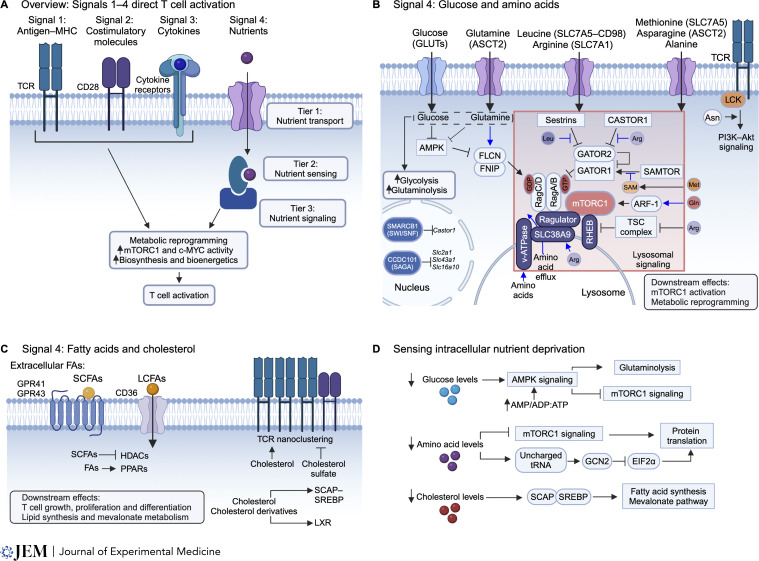
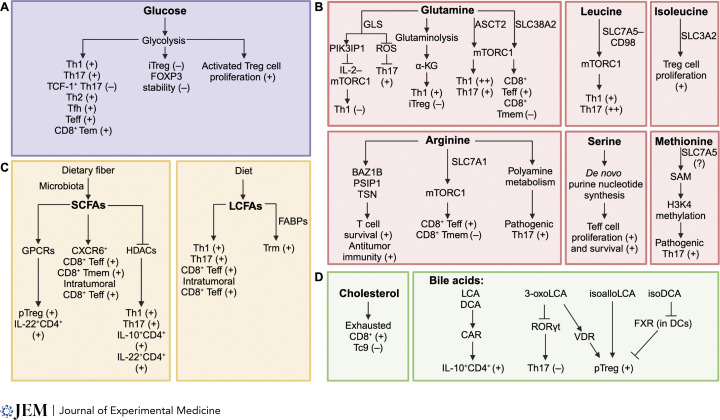
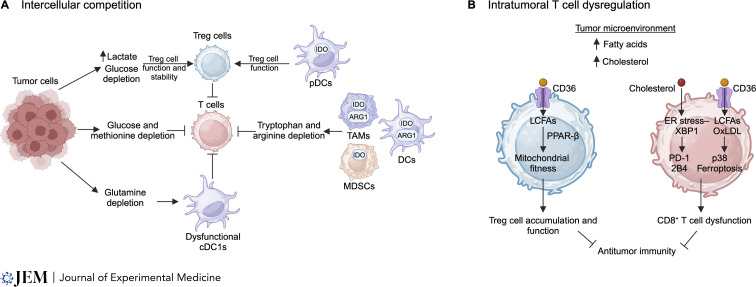
T cells are integral in mediating adaptive immunity to infection, autoimmunity, and cancer. Upon immune challenge, T cells exit from a quiescent state, followed by clonal expansion and effector differentiation. These processes are shaped by three established immune signals, namely antigen stimulation (Signal 1), costimulation (Signal 2), and cytokines (Signal 3). Emerging findings reveal that nutrients, including glucose, amino acids, and lipids, are crucial regulators of T cell responses and interplay with Signals 1-3, highlighting nutrients as Signal 4 to license T cell immunity. Here, we first summarize the functional importance of Signal 4 and the underlying mechanisms of nutrient transport, sensing, and signaling in orchestrating T cell activation and quiescence exit. We also discuss the roles of nutrients in programming T cell differentiation and functional fitness and how nutrients can be targeted to improve disease therapy. Understanding how T cells respond to Signal 4 nutrients in microenvironments will provide insights into context-dependent functions of adaptive immunity and therapeutic interventions.
© 2024 Raynor and Chi.
Conflict of interest statement
Disclosures: H. Chi reported personal fees from Kumquat Biosciences, Inc., Chugai Pharmaceuticals, and ONO Pharmaceutical Co. outside the submitted work; in addition, H. Chi has patents/patent applications in the field of immunotherapy pending. No other disclosures were reported.
Figures

Similar articles
-
Principles and therapeutic applications of adaptive immunity.Cell. 2024 Apr 25;187(9):2052-2078. doi: 10.1016/j.cell.2024.03.037. Cell. 2024. PMID: 38670065 Free PMC article. Review.
-
Metabolic coordination of T cell quiescence and activation.Nat Rev Immunol. 2020 Jan;20(1):55-70. doi: 10.1038/s41577-019-0203-y. Epub 2019 Aug 12. Nat Rev Immunol. 2020. PMID: 31406325 Review.
-
mTOR signaling in T cell immunity and autoimmunity.Int Rev Immunol. 2015 Jan;34(1):50-66. doi: 10.3109/08830185.2014.933957. Epub 2014 Jul 14. Int Rev Immunol. 2015. PMID: 25019278 Review.
-
Hallmarks of T-cell Exit from Quiescence.Cancer Immunol Res. 2018 May;6(5):502-508. doi: 10.1158/2326-6066.CIR-17-0605. Cancer Immunol Res. 2018. PMID: 29716982 Review.
-
T cell exit from quiescence and differentiation into Th2 cells depend on Raptor-mTORC1-mediated metabolic reprogramming.Immunity. 2013 Dec 12;39(6):1043-56. doi: 10.1016/j.immuni.2013.09.015. Epub 2013 Dec 5. Immunity. 2013. PMID: 24315998 Free PMC article.
Cited by
-
T cell dysfunction and therapeutic intervention in cancer.Nat Immunol. 2024 Aug;25(8):1344-1354. doi: 10.1038/s41590-024-01896-9. Epub 2024 Jul 18. Nat Immunol. 2024. PMID: 39025962 Free PMC article. Review.
-
The Nuclear Receptor NR1B1/RARα Arrests the Differentiation of Anti-Tumor Effector Cytotoxic T Cells.Adv Sci (Weinh). 2025 May;12(17):e2410241. doi: 10.1002/advs.202410241. Epub 2025 Mar 11. Adv Sci (Weinh). 2025. PMID: 40068101 Free PMC article.
-
Principles and therapeutic applications of adaptive immunity.Cell. 2024 Apr 25;187(9):2052-2078. doi: 10.1016/j.cell.2024.03.037. Cell. 2024. PMID: 38670065 Free PMC article. Review.
-
Architects of immunity: How dendritic cells shape CD8+ T cell fate in cancer.Sci Immunol. 2025 Jan 17;10(103):eadf4726. doi: 10.1126/sciimmunol.adf4726. Epub 2025 Jan 17. Sci Immunol. 2025. PMID: 39823318 Free PMC article. Review.
-
Targeting metabolic dysfunction of CD8 T cells and natural killer cells in cancer.Nat Rev Drug Discov. 2025 Mar;24(3):190-208. doi: 10.1038/s41573-024-01098-w. Epub 2024 Dec 12. Nat Rev Drug Discov. 2025. PMID: 39668206 Review.
References
-
- Alizadeh, D., Wong R.A., Yang X., Wang D., Pecoraro J.R., Kuo C.F., Aguilar B., Qi Y., Ann D.K., Starr R., et al. . 2019. IL15 enhances CAR-T cell antitumor activity by reducing mTORC1 activity and preserving their stem cell memory phenotype. Cancer Immunol. Res. 7:759–772. 10.1158/2326-6066.CIR-18-0466 - DOI - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
