

SNUPN deficiency causes a recessive muscular dystrophy due to RNA mis-splicing and ECM dysregulation
- PMID: 38413582
- PMCID: PMC10899626
- DOI: 10.1038/s41467-024-45933-5
SNUPN deficiency causes a recessive muscular dystrophy due to RNA mis-splicing and ECM dysregulation
Abstract
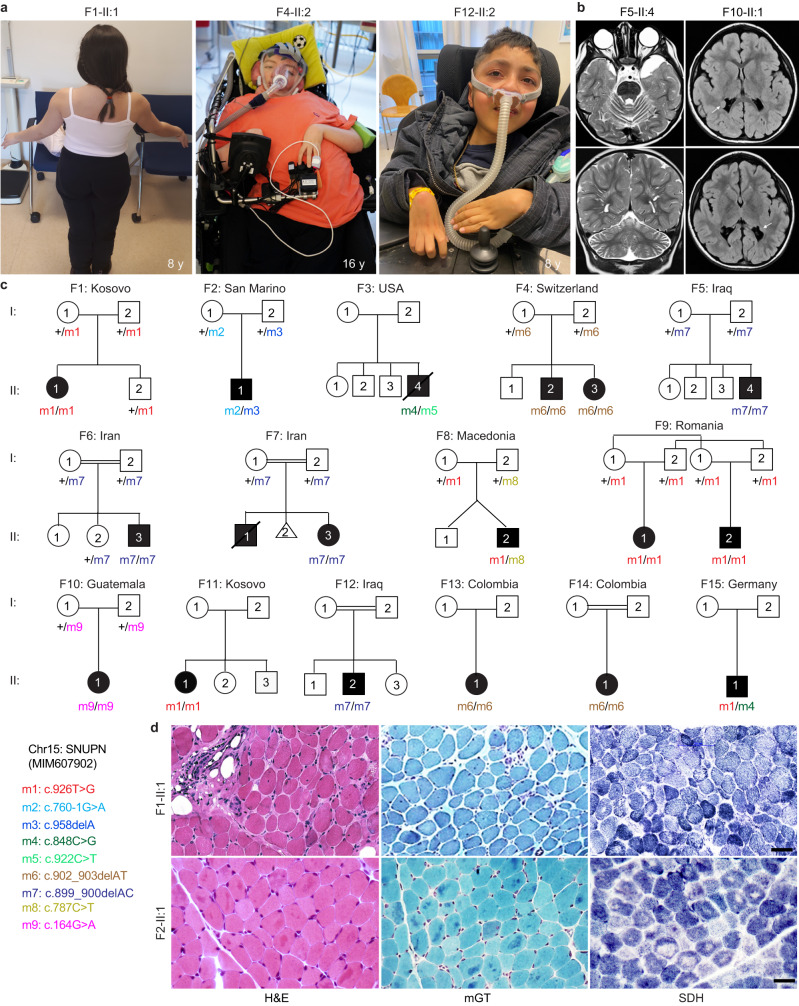
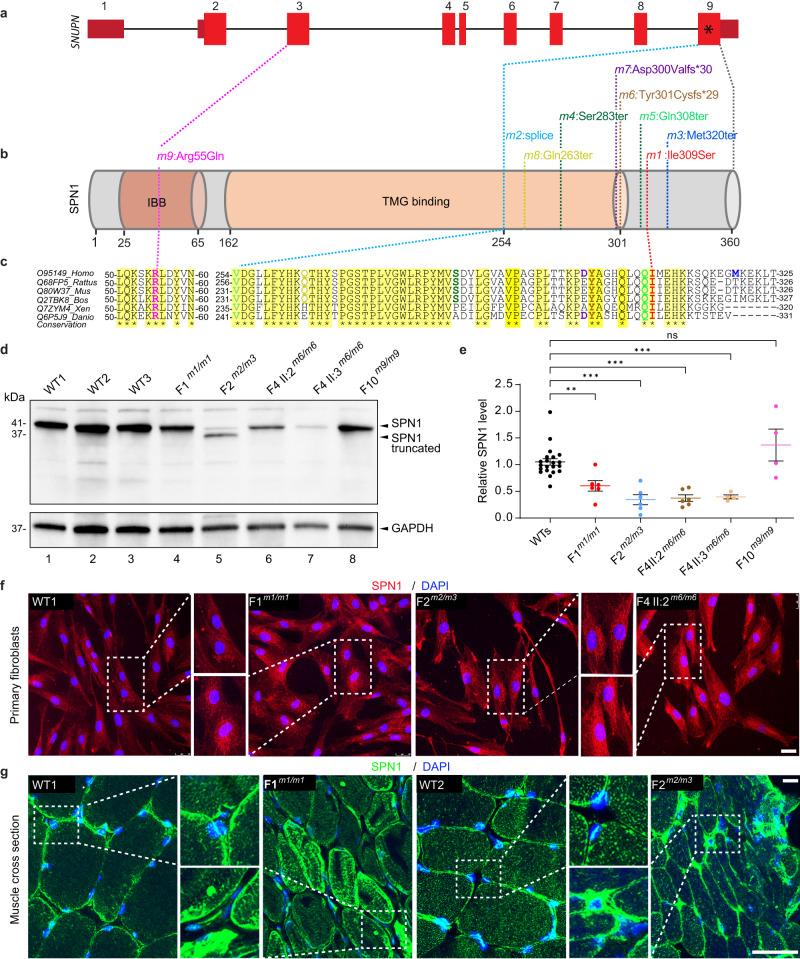
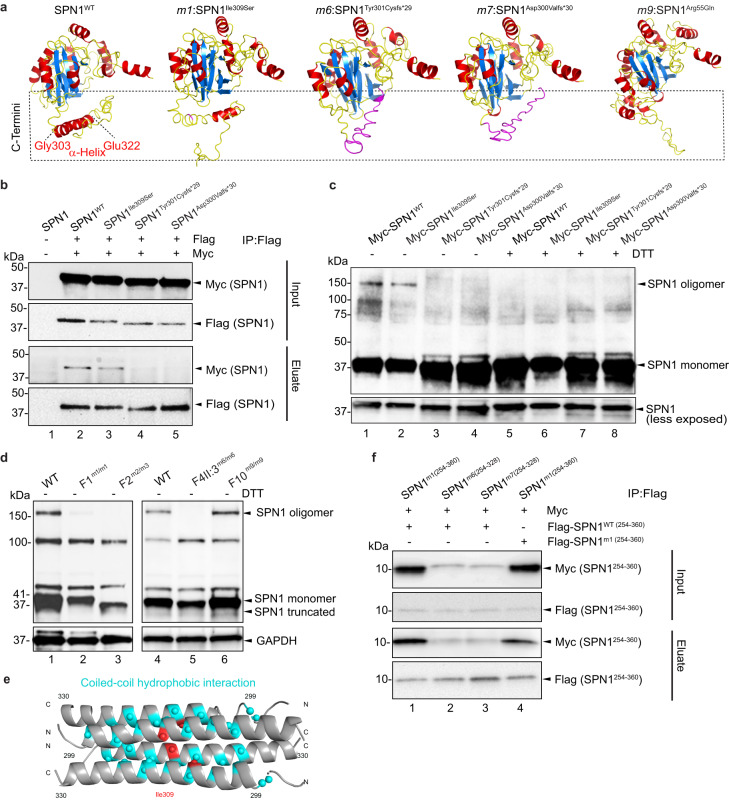
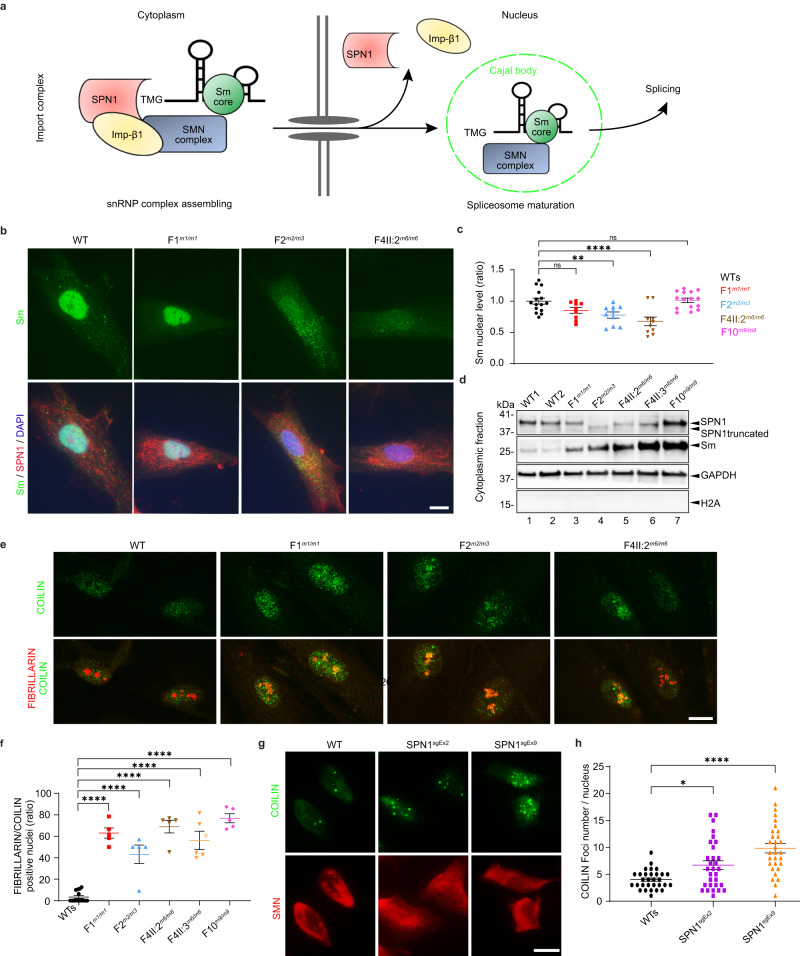
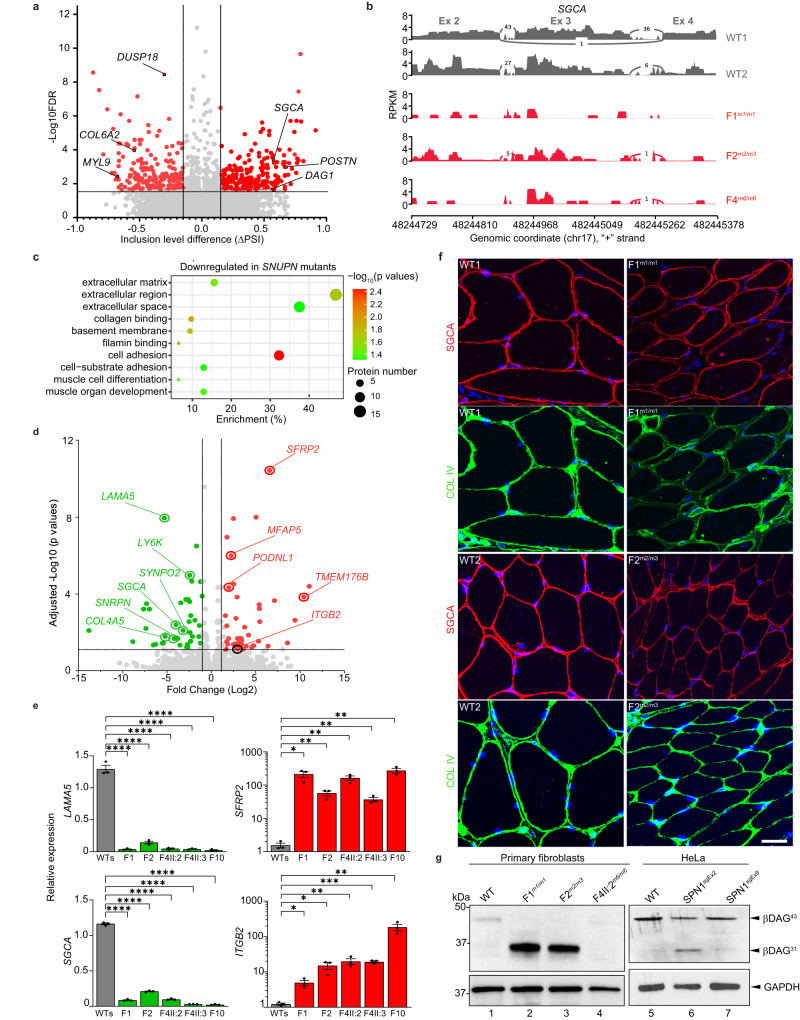
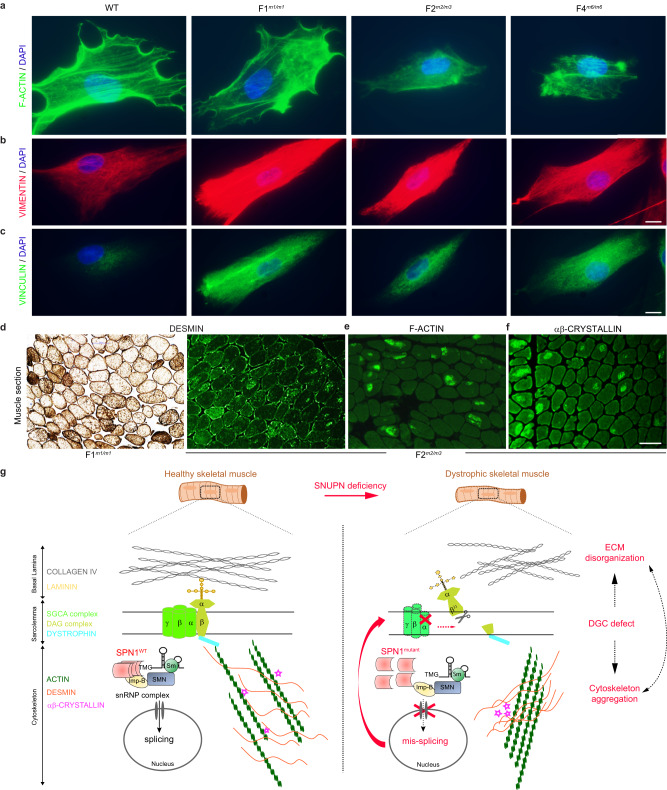
SNURPORTIN-1, encoded by SNUPN, plays a central role in the nuclear import of spliceosomal small nuclear ribonucleoproteins. However, its physiological function remains unexplored. In this study, we investigate 18 children from 15 unrelated families who present with atypical muscular dystrophy and neurological defects. Nine hypomorphic SNUPN biallelic variants, predominantly clustered in the last coding exon, are ascertained to segregate with the disease. We demonstrate that mutant SPN1 failed to oligomerize leading to cytoplasmic aggregation in patients' primary fibroblasts and CRISPR/Cas9-mediated mutant cell lines. Additionally, mutant nuclei exhibit defective spliceosomal maturation and breakdown of Cajal bodies. Transcriptome analyses reveal splicing and mRNA expression dysregulation, particularly in sarcolemmal components, causing disruption of cytoskeletal organization in mutant cells and patient muscle tissues. Our findings establish SNUPN deficiency as the genetic etiology of a previously unrecognized subtype of muscular dystrophy and provide robust evidence of the role of SPN1 for muscle homeostasis.
© 2024. The Author(s).
Conflict of interest statement
M.E.R., S.K., A.P. and A.B.A. are employed by and receive a salary from Centogene AG. The remaining authors declare no competing interests.
Figures
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
