

Combining full-length gene assay and SpliceAI to interpret the splicing impact of all possible SPINK1 coding variants
- PMID: 38414044
- PMCID: PMC10898081
- DOI: 10.1186/s40246-024-00586-9
Combining full-length gene assay and SpliceAI to interpret the splicing impact of all possible SPINK1 coding variants
Abstract
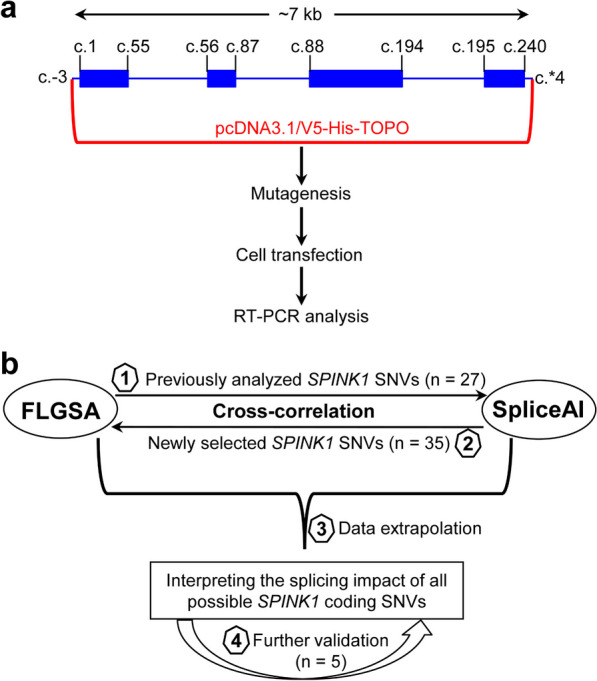
Background: Single-nucleotide variants (SNVs) within gene coding sequences can significantly impact pre-mRNA splicing, bearing profound implications for pathogenic mechanisms and precision medicine. In this study, we aim to harness the well-established full-length gene splicing assay (FLGSA) in conjunction with SpliceAI to prospectively interpret the splicing effects of all potential coding SNVs within the four-exon SPINK1 gene, a gene associated with chronic pancreatitis.
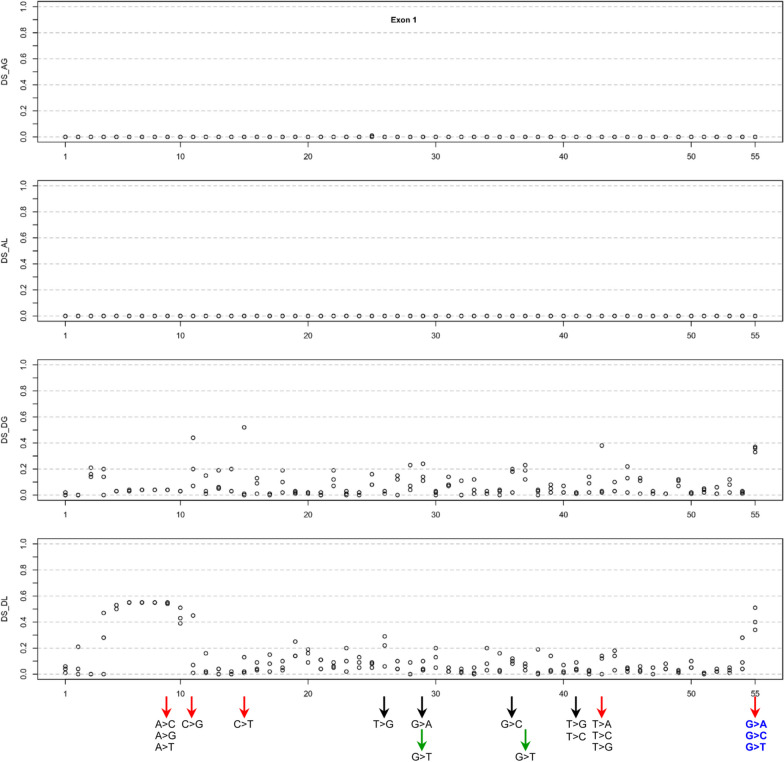
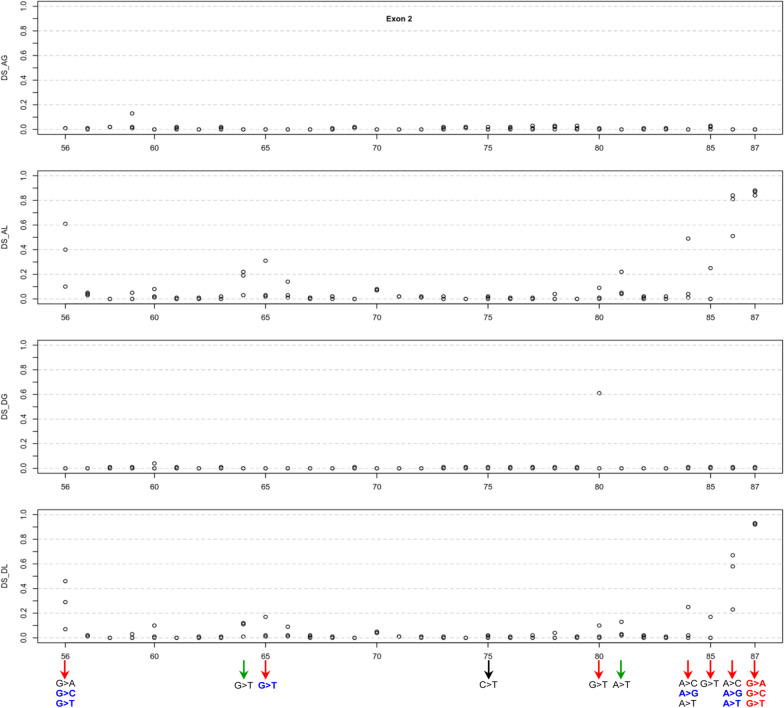
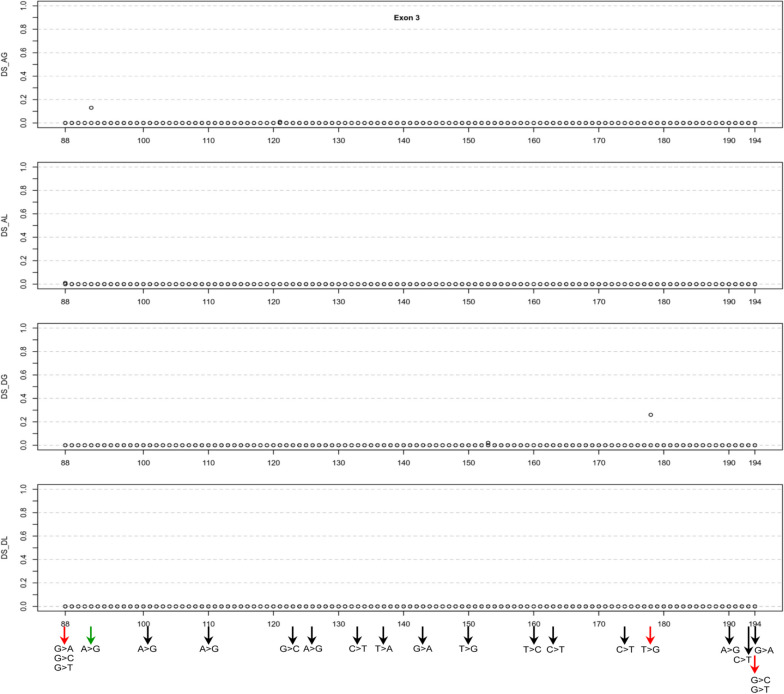
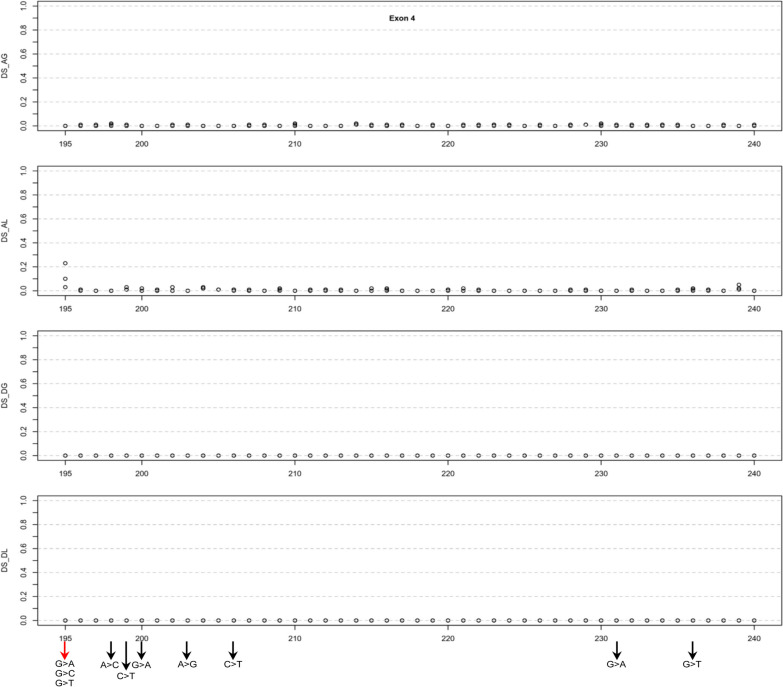
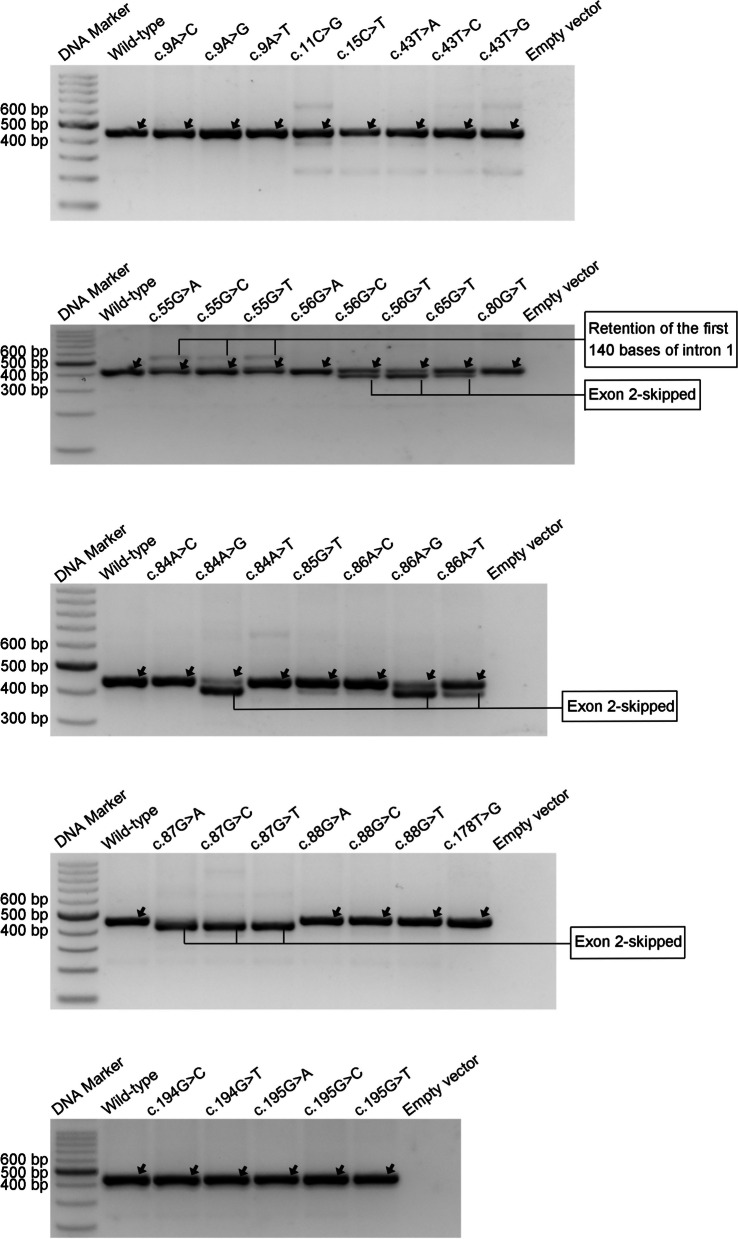
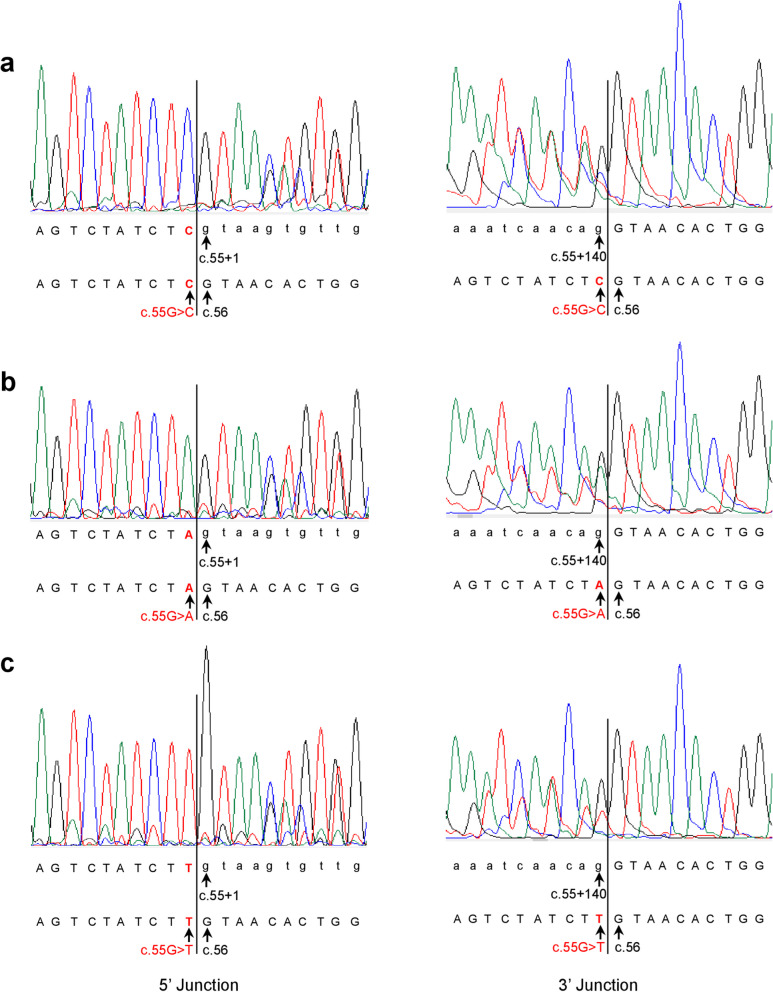
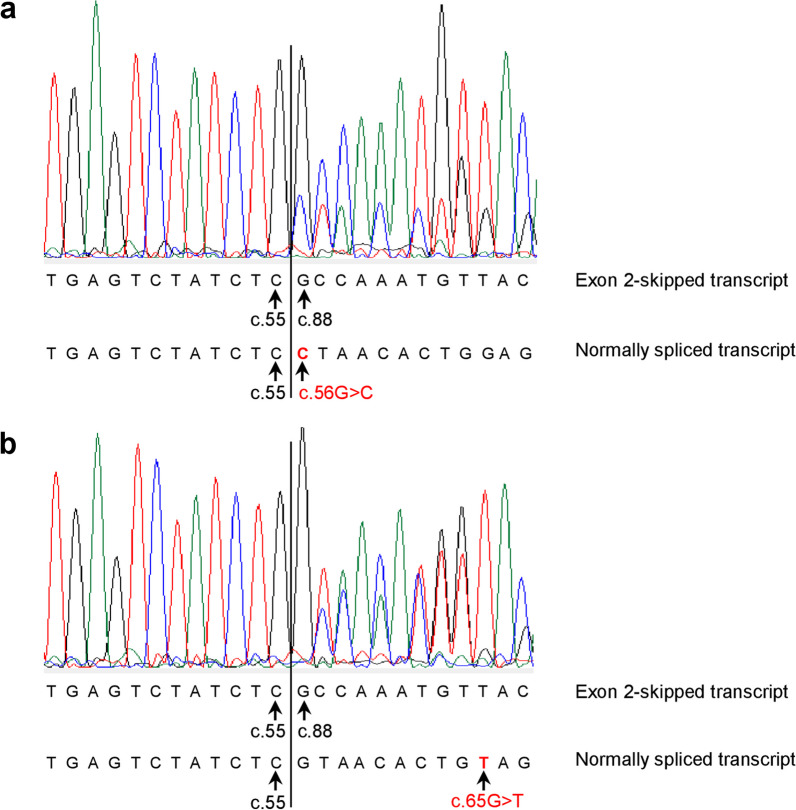
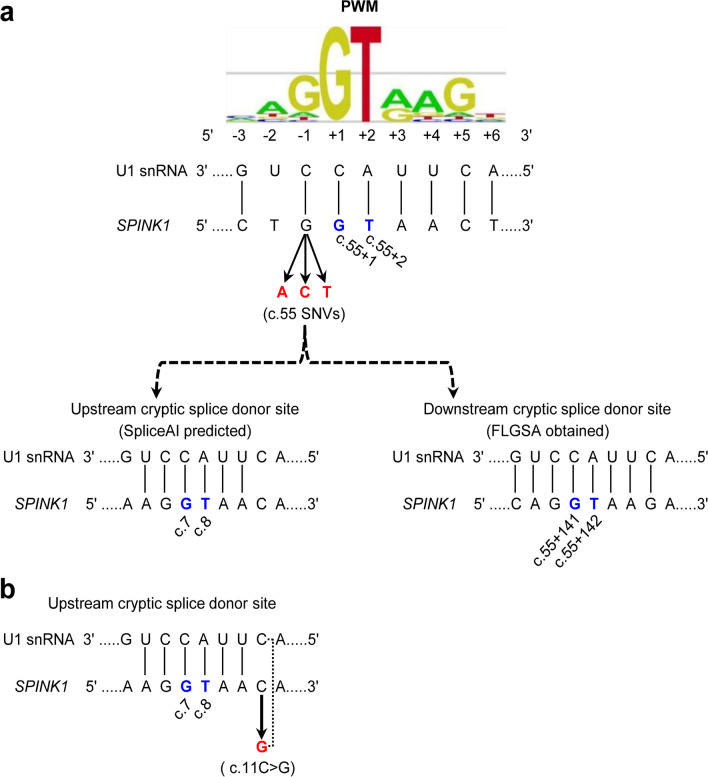
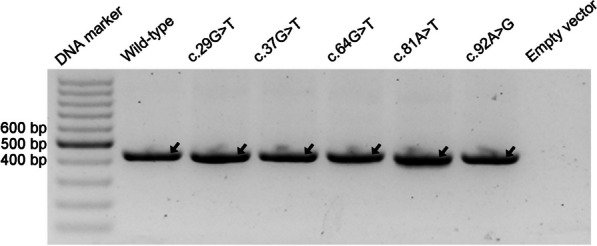
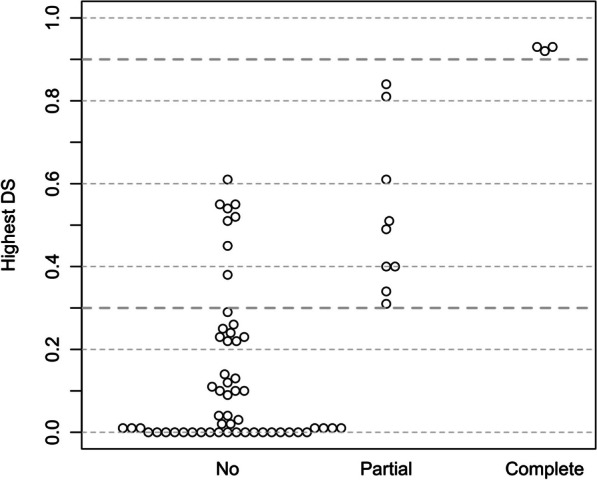
Results: Our study began with a retrospective analysis of 27 SPINK1 coding SNVs previously assessed using FLGSA, proceeded with a prospective analysis of 35 new FLGSA-tested SPINK1 coding SNVs, followed by data extrapolation, and ended with further validation. In total, we analyzed 67 SPINK1 coding SNVs, which account for 9.3% of the 720 possible coding SNVs. Among these 67 FLGSA-analyzed SNVs, 12 were found to impact splicing. Through detailed comparison of FLGSA results and SpliceAI predictions, we inferred that the remaining 653 untested coding SNVs in the SPINK1 gene are unlikely to significantly affect splicing. Of the 12 splice-altering events, nine produced both normally spliced and aberrantly spliced transcripts, while the remaining three only generated aberrantly spliced transcripts. These splice-impacting SNVs were found solely in exons 1 and 2, notably at the first and/or last coding nucleotides of these exons. Among the 12 splice-altering events, 11 were missense variants (2.17% of 506 potential missense variants), and one was synonymous (0.61% of 164 potential synonymous variants). Notably, adjusting the SpliceAI cut-off to 0.30 instead of the conventional 0.20 would improve specificity without reducing sensitivity.
Conclusions: By integrating FLGSA with SpliceAI, we have determined that less than 2% (1.67%) of all possible coding SNVs in SPINK1 significantly influence splicing outcomes. Our findings emphasize the critical importance of conducting splicing analysis within the broader genomic sequence context of the study gene and highlight the inherent uncertainties associated with intermediate SpliceAI scores (0.20 to 0.80). This study contributes to the field by being the first to prospectively interpret all potential coding SNVs in a disease-associated gene with a high degree of accuracy, representing a meaningful attempt at shifting from retrospective to prospective variant analysis in the era of exome and genome sequencing.
Keywords: SPINK1 gene; Chronic pancreatitis; Full-length gene splicing assay (FLGSA); Pre-mRNA splicing; Precision medicine in genetics; Single-nucleotide variants (SNVs); Splice site; SpliceAI; Splicing prediction algorithms; Variant interpretation.
© 2024. The Author(s).
Conflict of interest statement
Jian-Min Chen serves as an Associate Editor for Human Genomics but was not involved in the editorial review process or the decision to publish this article. All remaining authors declare that they have no competing interests.
Figures
Similar articles
-
Genetics and clinical implications of SPINK1 in the pancreatitis continuum and pancreatic cancer.Hum Genomics. 2025 Mar 26;19(1):32. doi: 10.1186/s40246-025-00740-x. Hum Genomics. 2025. PMID: 40140953 Free PMC article. Review.
-
Toward a clinical diagnostic pipeline for SPINK1 intronic variants.Hum Genomics. 2019 Feb 12;13(1):8. doi: 10.1186/s40246-019-0193-7. Hum Genomics. 2019. PMID: 30755276 Free PMC article.
-
Exploring the role of splicing in TP53 variant pathogenicity through predictions and minigene assays.Hum Genomics. 2025 Jan 8;19(1):2. doi: 10.1186/s40246-024-00714-5. Hum Genomics. 2025. PMID: 39780207 Free PMC article.
-
Splicing Outcomes of 5' Splice Site GT>GC Variants That Generate Wild-Type Transcripts Differ Significantly Between Full-Length and Minigene Splicing Assays.Front Genet. 2021 Aug 5;12:701652. doi: 10.3389/fgene.2021.701652. eCollection 2021. Front Genet. 2021. PMID: 34422003 Free PMC article.
-
Clinical interpretation of SPINK1 and CTRC variants in pancreatitis.Pancreatology. 2020 Oct;20(7):1354-1367. doi: 10.1016/j.pan.2020.09.001. Epub 2020 Sep 7. Pancreatology. 2020. PMID: 32948427
Cited by
-
Alu insertion-mediated dsRNA structure formation with pre-existing Alu elements as a disease-causing mechanism.Am J Hum Genet. 2024 Oct 3;111(10):2176-2189. doi: 10.1016/j.ajhg.2024.08.016. Epub 2024 Sep 11. Am J Hum Genet. 2024. PMID: 39265574 Free PMC article.
-
Genetics and clinical implications of SPINK1 in the pancreatitis continuum and pancreatic cancer.Hum Genomics. 2025 Mar 26;19(1):32. doi: 10.1186/s40246-025-00740-x. Hum Genomics. 2025. PMID: 40140953 Free PMC article. Review.
-
U-rich elements drive pervasive cryptic splicing in 3' UTR massively parallel reporter assays.Nat Commun. 2025 Jul 25;16(1):6844. doi: 10.1038/s41467-025-62000-9. Nat Commun. 2025. PMID: 40715118 Free PMC article.
-
U-rich elements drive pervasive cryptic splicing in 3' UTR massively parallel reporter assays.bioRxiv [Preprint]. 2024 Aug 5:2024.08.05.606557. doi: 10.1101/2024.08.05.606557. bioRxiv. 2024. Update in: Nat Commun. 2025 Jul 25;16(1):6844. doi: 10.1038/s41467-025-62000-9. PMID: 39149310 Free PMC article. Updated. Preprint.
References
-
- Cartegni L, Chew SL, Krainer AR. Listening to silence and understanding nonsense: exonic mutations that affect splicing. Nat Rev Genet. 2002;3(4):285–298. - PubMed
-
- Sarkar A, Panati K, Narala VR. Code inside the codon: The role of synonymous mutations in regulating splicing machinery and its impact on disease. Mutat Res Rev Mutat Res. 2022;790:108444. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
