

Acute Adenoviral Infection Elicits an Arrhythmogenic Substrate Prior to Myocarditis
- PMID: 38415360
- PMCID: PMC11003857
- DOI: 10.1161/CIRCRESAHA.122.322437
Acute Adenoviral Infection Elicits an Arrhythmogenic Substrate Prior to Myocarditis
Abstract
Background: Viral cardiac infection represents a significant clinical challenge encompassing several etiological agents, disease stages, complex presentation, and a resulting lack of mechanistic understanding. Myocarditis is a major cause of sudden cardiac death in young adults, where current knowledge in the field is dominated by later disease phases and pathological immune responses. However, little is known regarding how infection can acutely induce an arrhythmogenic substrate before significant immune responses. Adenovirus is a leading cause of myocarditis, but due to species specificity, models of infection are lacking, and it is not understood how adenoviral infection may underlie sudden cardiac arrest. Mouse adenovirus type-3 was previously reported as cardiotropic, yet it has not been utilized to understand the mechanisms of cardiac infection and pathology.
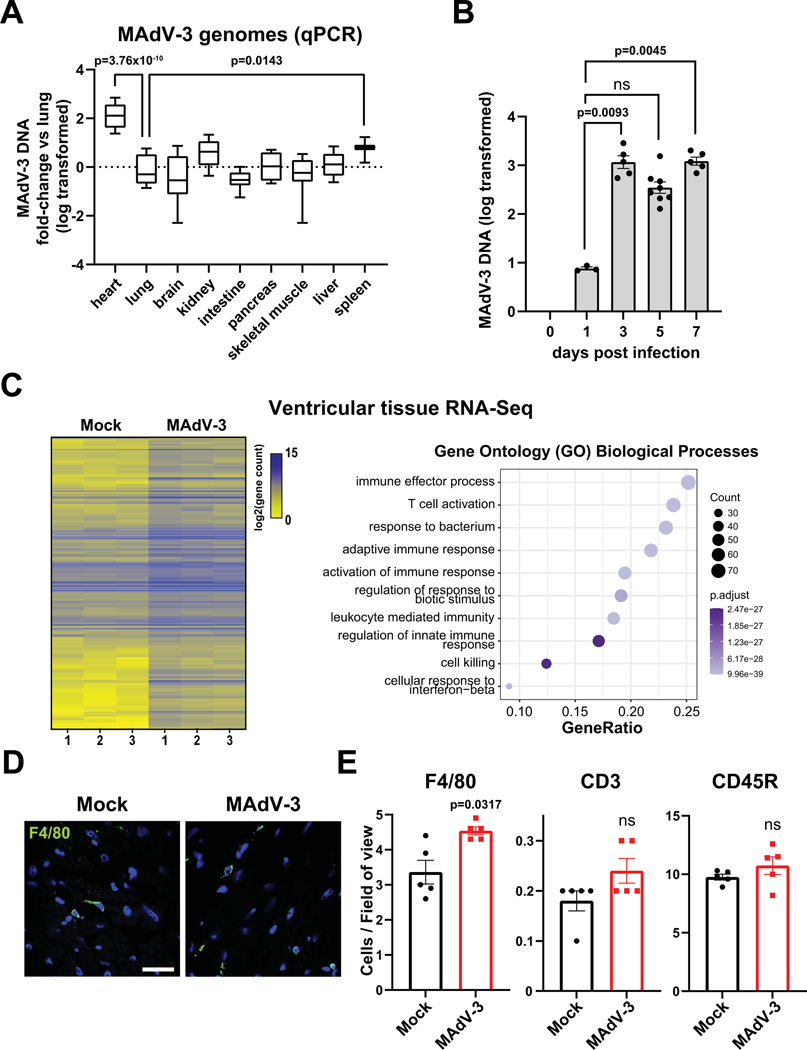
Methods: We have developed mouse adenovirus type-3 infection as a model to investigate acute cardiac infection and molecular alterations to the infected heart before an appreciable immune response or gross cardiomyopathy.
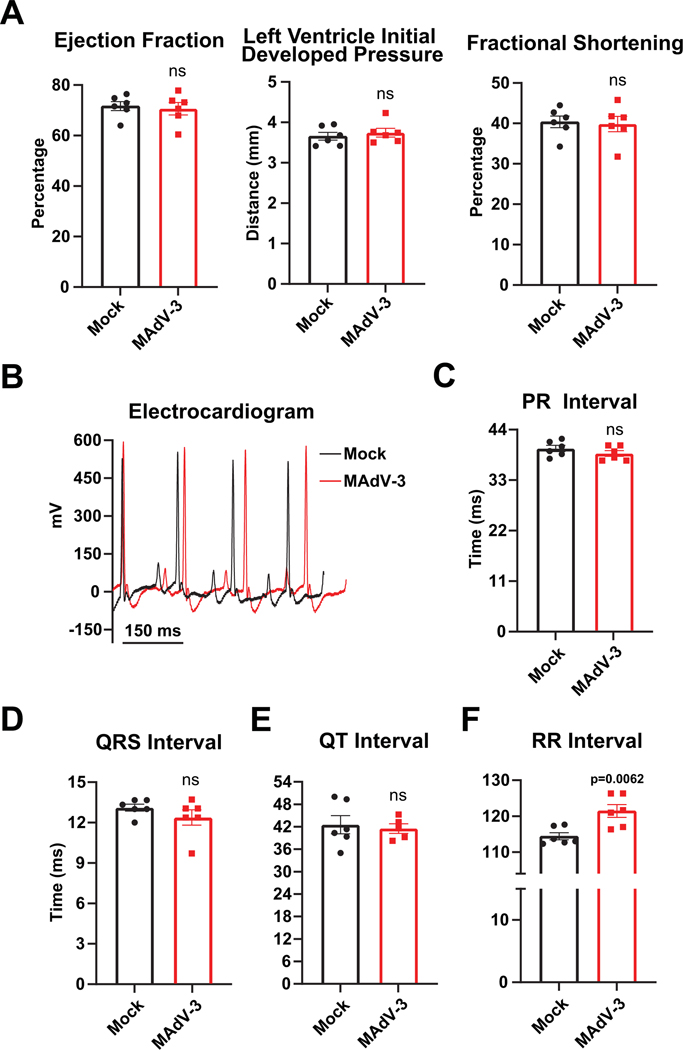
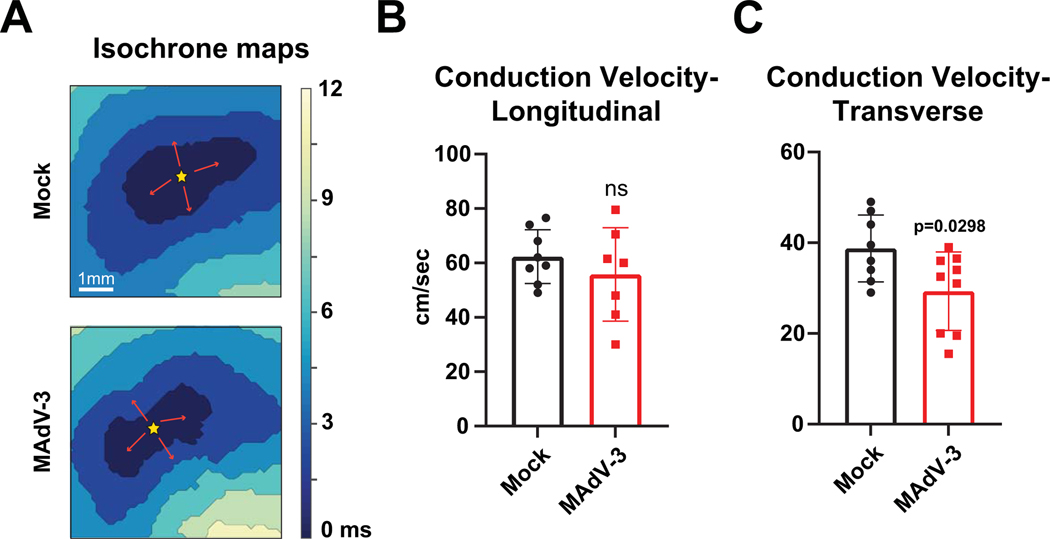
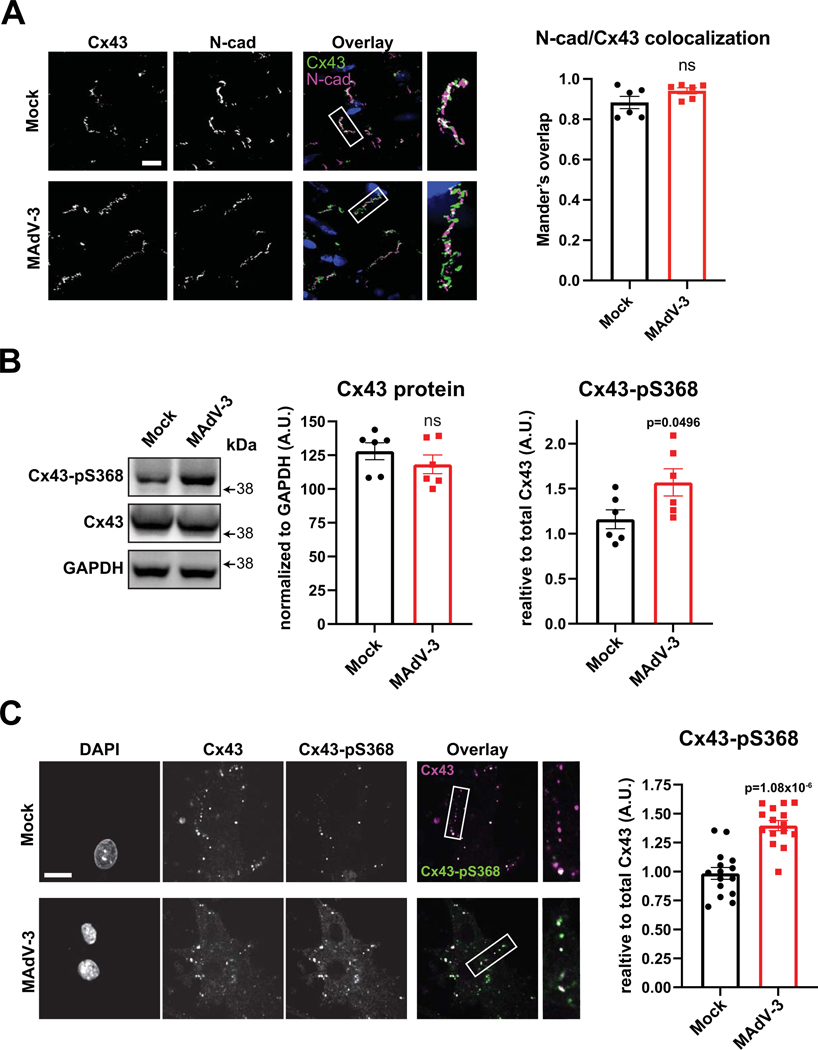
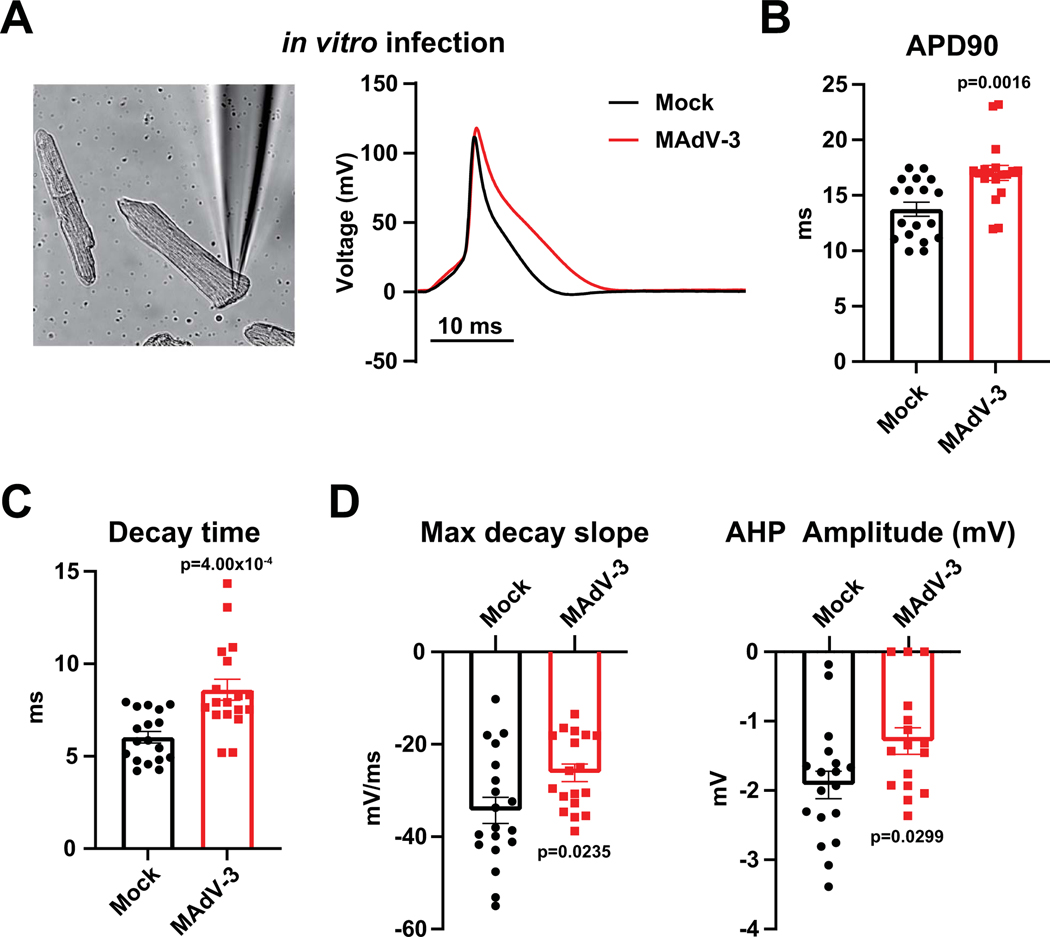
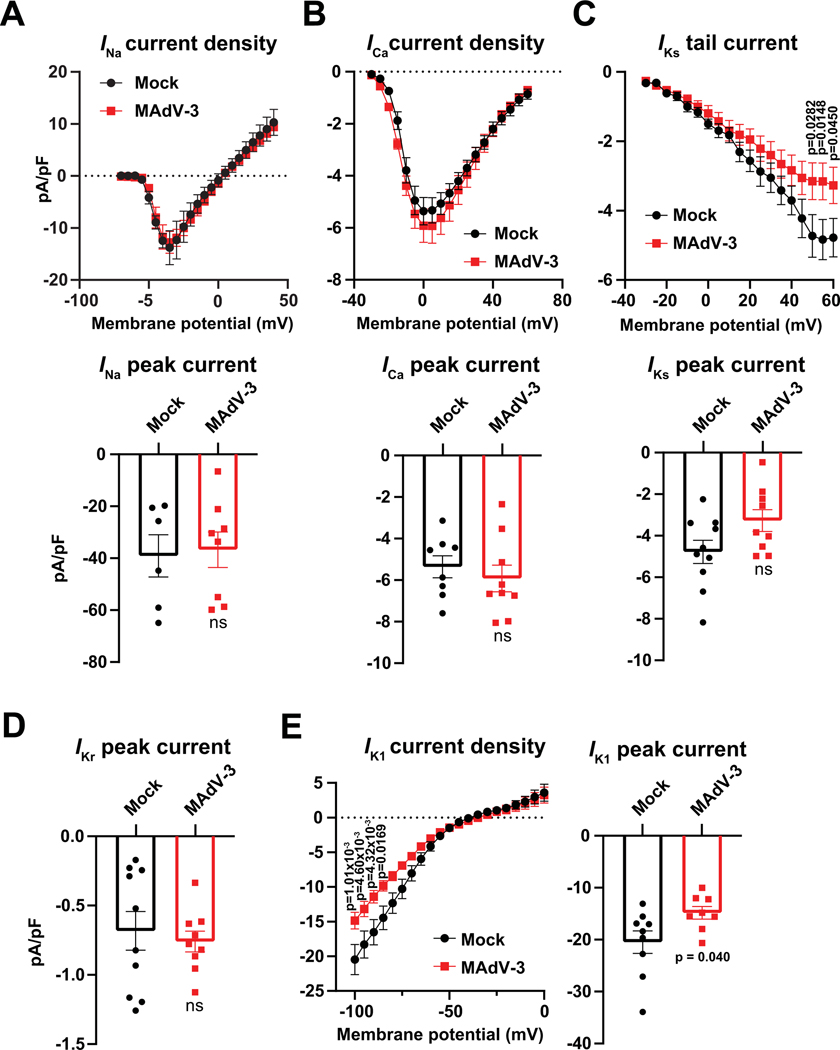
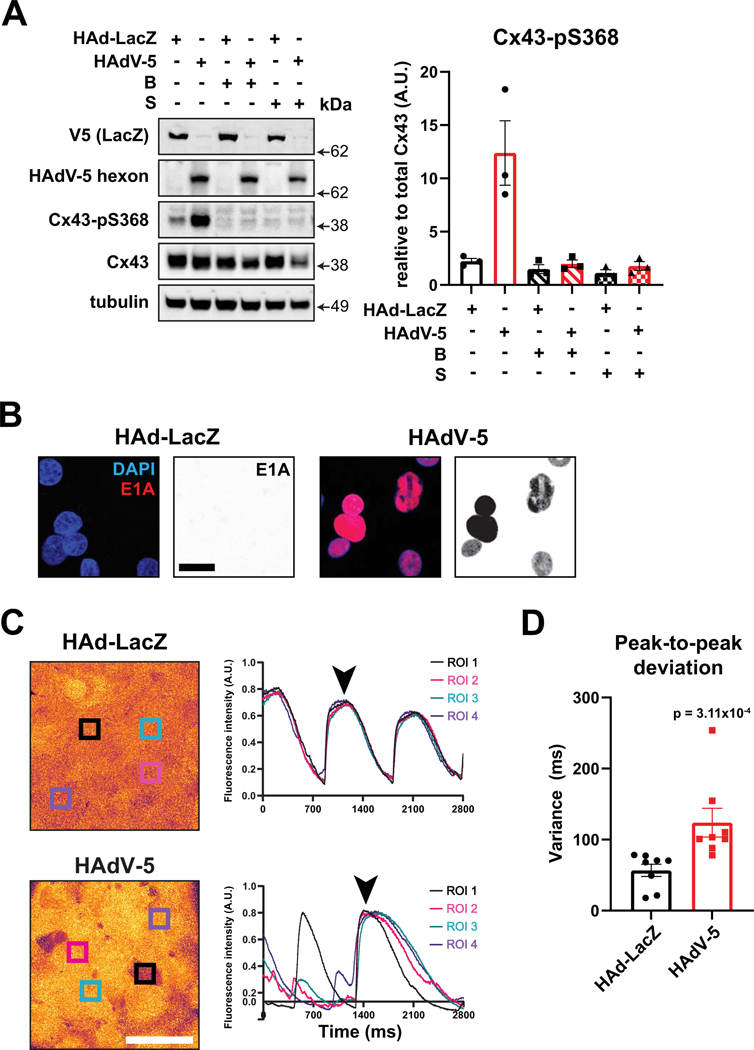
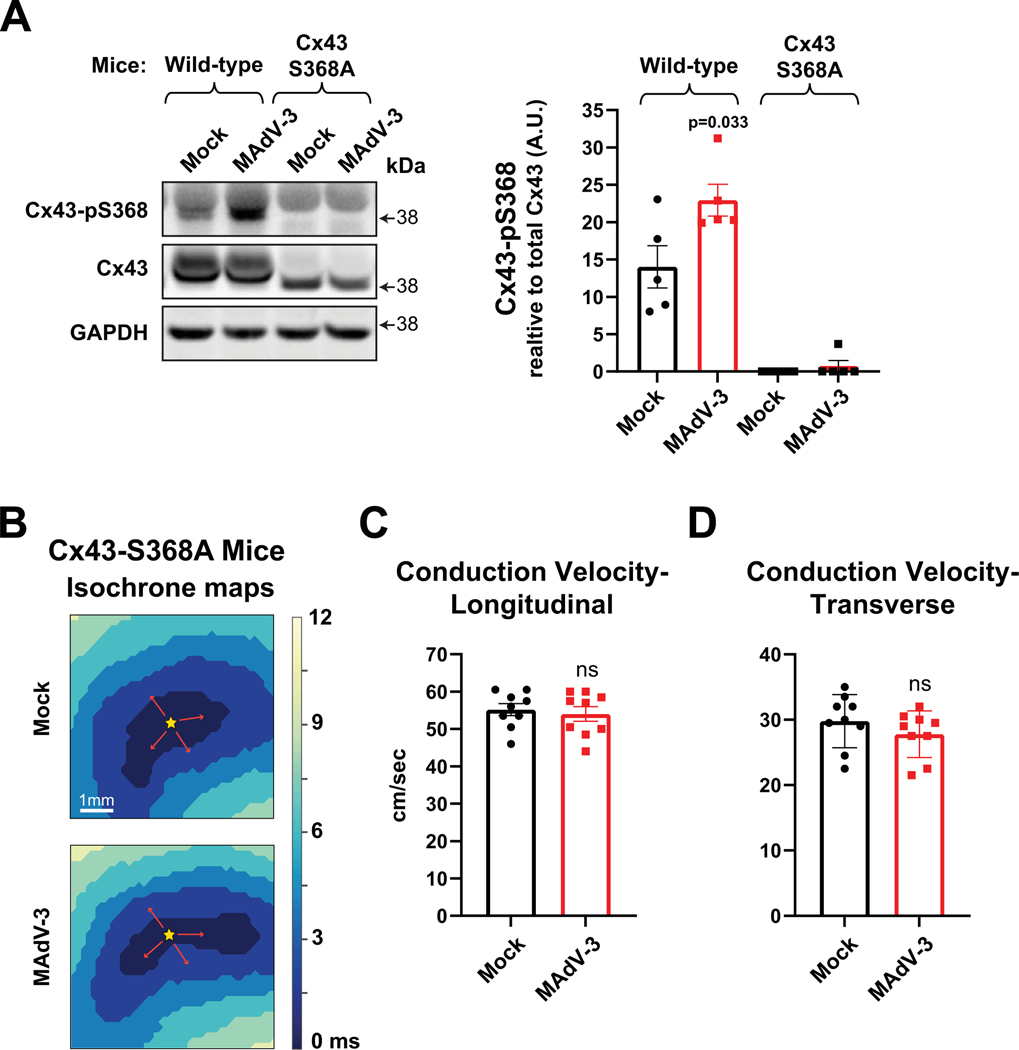
Results: Optical mapping of infected hearts exposes decreases in conduction velocity concomitant with increased Cx43Ser368 phosphorylation, a residue known to regulate gap junction function. Hearts from animals harboring a phospho-null mutation at Cx43Ser368 are protected against mouse adenovirus type-3-induced conduction velocity slowing. Additional to gap junction alterations, patch clamping of mouse adenovirus type-3-infected adult mouse ventricular cardiomyocytes reveals prolonged action potential duration as a result of decreased IK1 and IKs current density. Turning to human systems, we find human adenovirus type-5 increases phosphorylation of Cx43Ser368 and disrupts synchrony in human induced pluripotent stem cell-derived cardiomyocytes, indicating common mechanisms with our mouse whole heart and adult cardiomyocyte data.
Conclusions: Together, these findings demonstrate that adenoviral infection creates an arrhythmogenic substrate through direct targeting of gap junction and ion channel function in the heart. Such alterations are known to precipitate arrhythmias and likely contribute to sudden cardiac death in acutely infected patients.
Keywords: adenoviridae; arrhythmias, cardiac; death, sudden, cardiac; gap junctions; immunity; ion channels.
Conflict of interest statement
Figures
References
-
- Babu-Narayan SV, McCarthy KP, Ho SY, Magee AG, Kilner PJ, Sheppard MN. Images in cardiovascular medicine. Myocarditis and sudden cardiac death in the young: extensive fibrosis suggested by cardiovascular magnetic resonance in vivo and confirmed post mortem. Circulation. 2007;116:e122–125. doi: 10.1161/CIRCULATIONAHA.107.693085 - DOI - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
