

Loss of GTF2I promotes neuronal apoptosis and synaptic reduction in human cellular models of neurodevelopment
- PMID: 38416640
- PMCID: PMC11002531
- DOI: 10.1016/j.celrep.2024.113867
Loss of GTF2I promotes neuronal apoptosis and synaptic reduction in human cellular models of neurodevelopment
Abstract
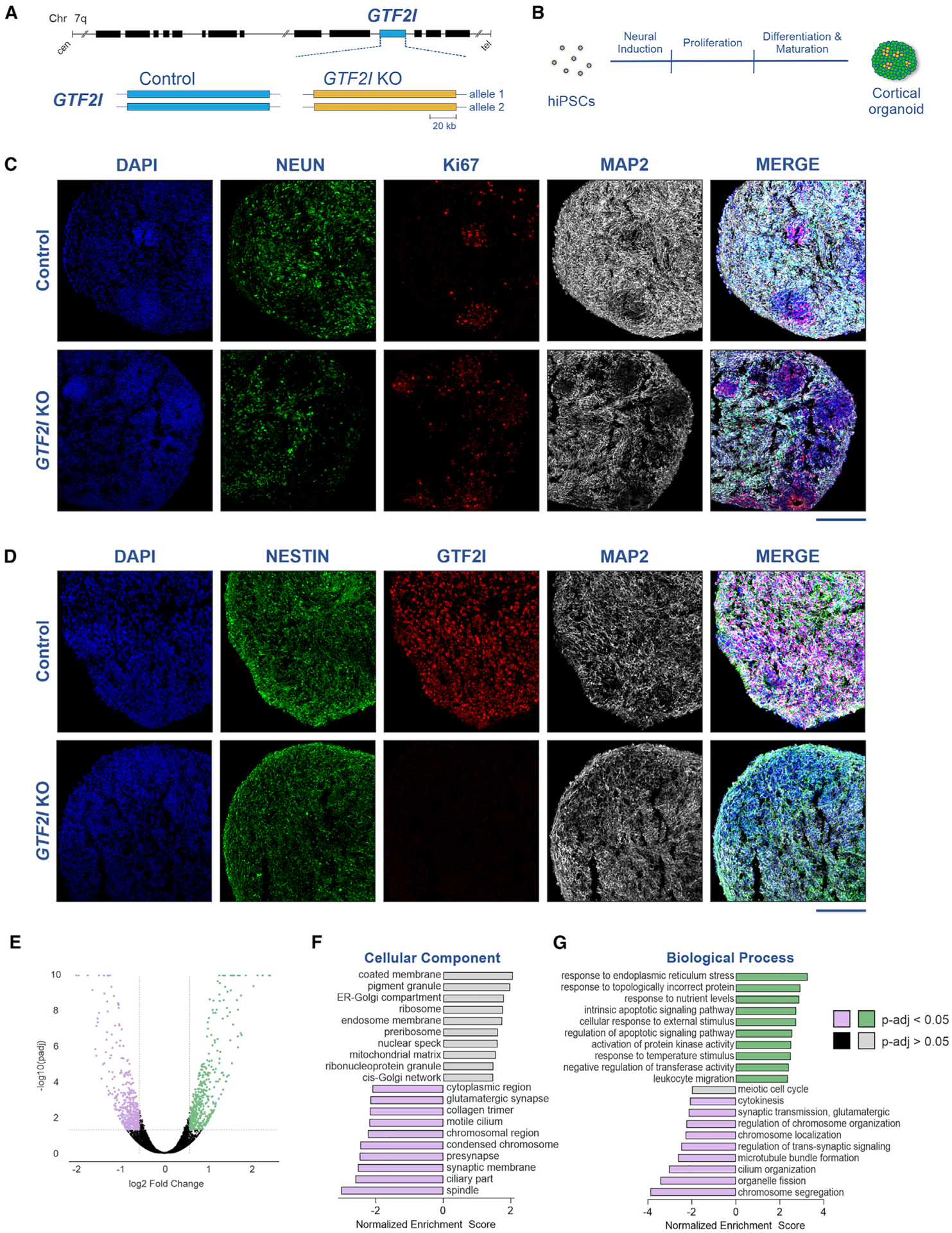
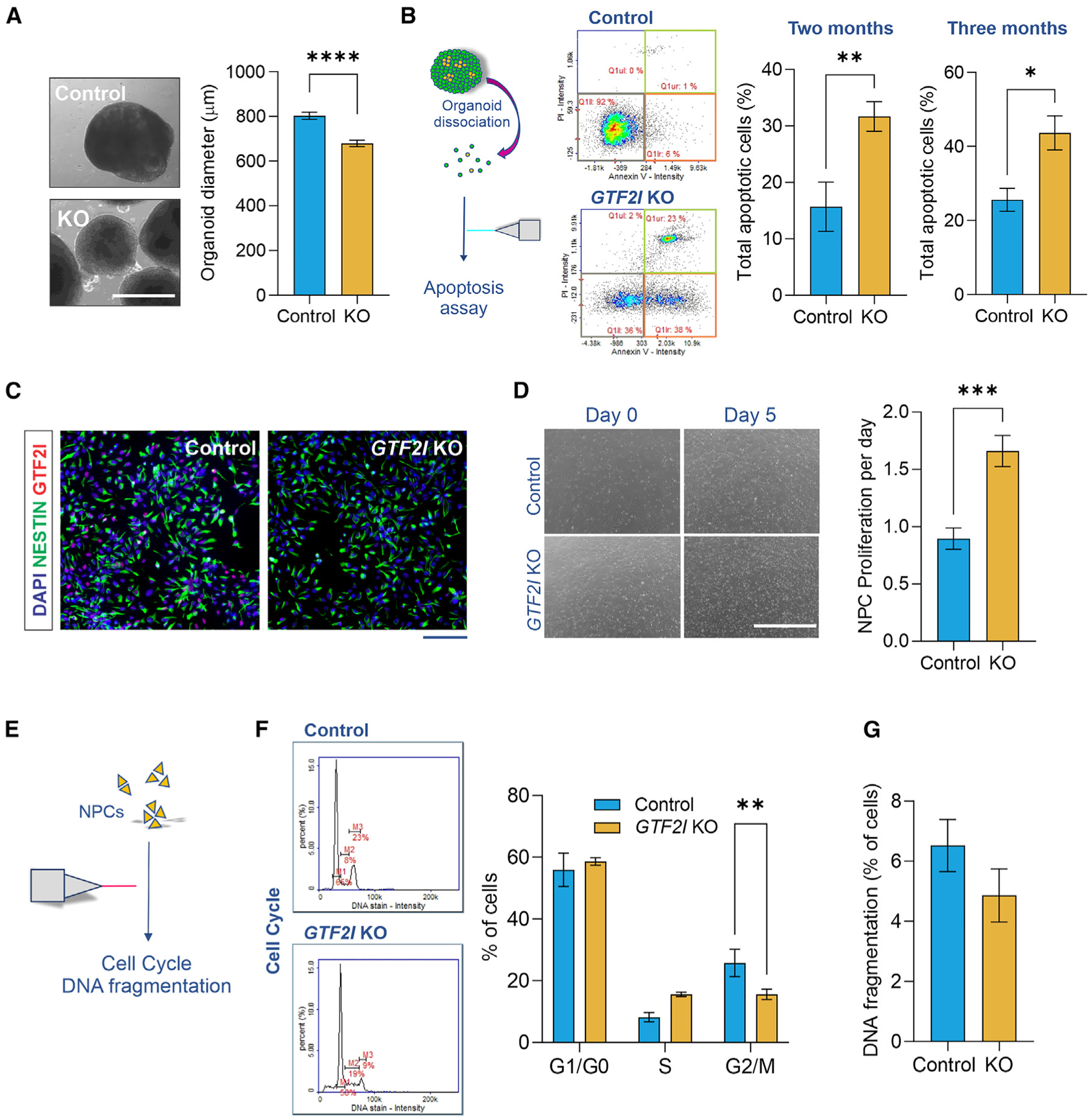
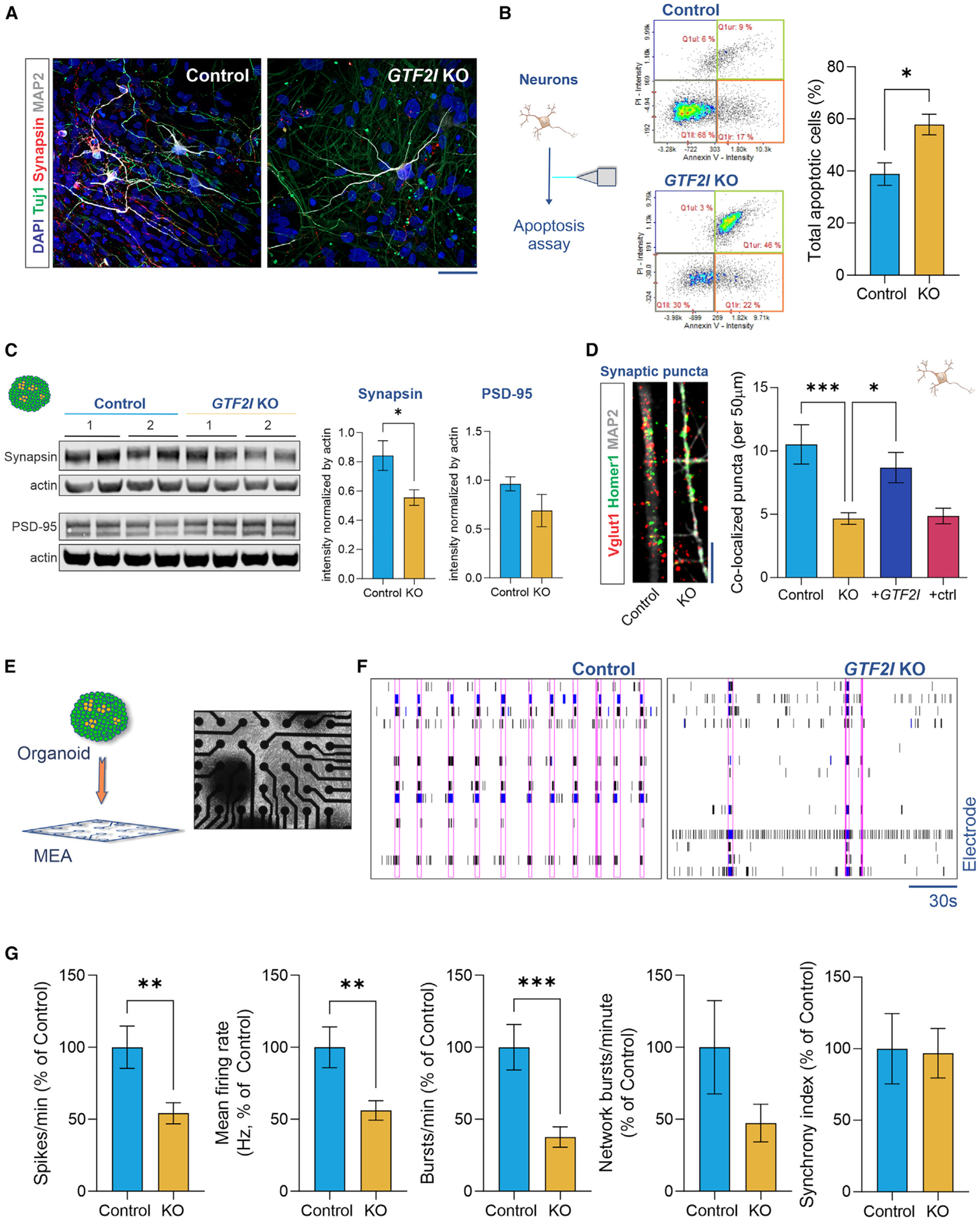
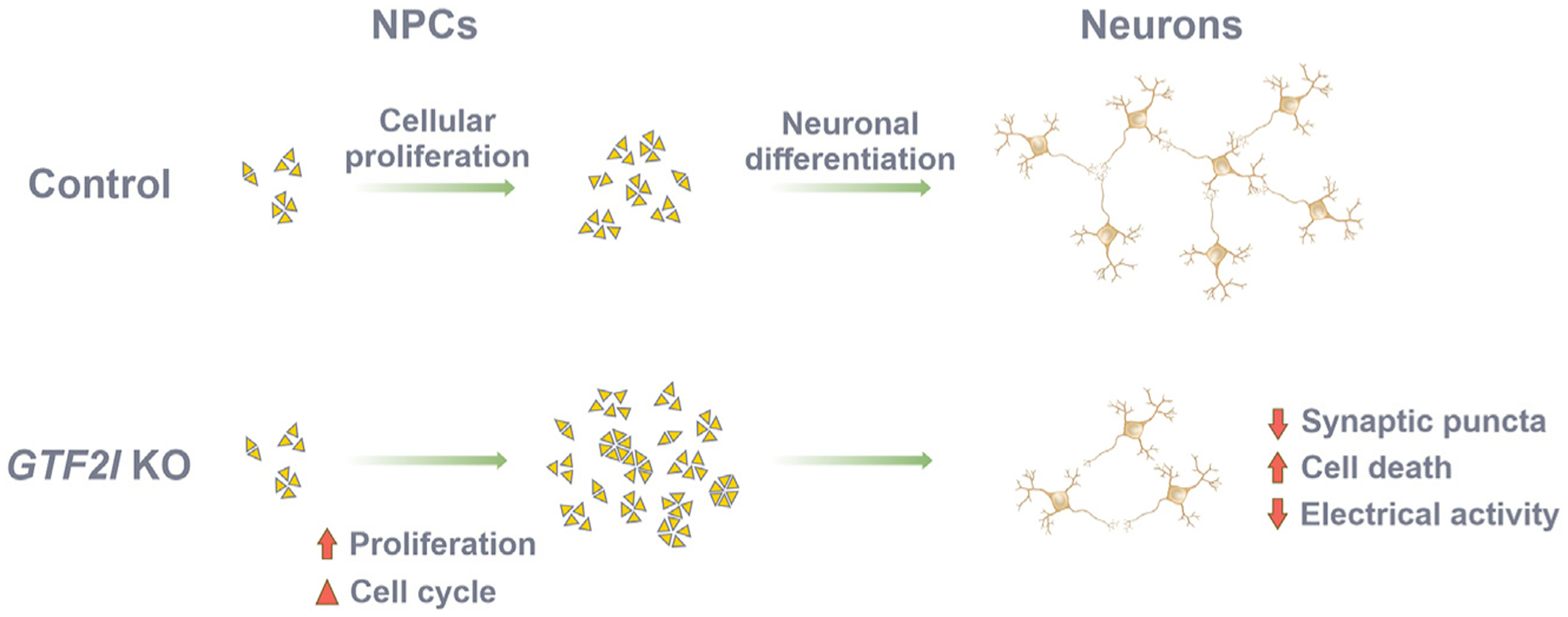
Individuals with Williams syndrome (WS), a neurodevelopmental disorder caused by hemizygous loss of 26-28 genes at 7q11.23, characteristically portray a hypersocial phenotype. Copy-number variations and mutations in one of these genes, GTF2I, are associated with altered sociality and are proposed to underlie hypersociality in WS. However, the contribution of GTF2I to human neurodevelopment remains poorly understood. Here, human cellular models of neurodevelopment, including neural progenitors, neurons, and three-dimensional cortical organoids, are differentiated from CRISPR-Cas9-edited GTF2I-knockout (GTF2I-KO) pluripotent stem cells to investigate the role of GTF2I in human neurodevelopment. GTF2I-KO progenitors exhibit increased proliferation and cell-cycle alterations. Cortical organoids and neurons demonstrate increased cell death and synaptic dysregulation, including synaptic structural dysfunction and decreased electrophysiological activity on a multielectrode array. Our findings suggest that changes in synaptic circuit integrity may be a prominent mediator of the link between alterations in GTF2I and variation in the phenotypic expression of human sociality.
Keywords: CP: Developmental biology; CP: Neuroscience; GTF2I; Williams syndrome; brain organoid; cortical organoid; neurodevelopment; stem cells.
Copyright © 2024 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests A.R.M. is a cofounder and has equity interest in TISMOO, a company dedicated to genetic analysis and brain organoid modeling focusing on therapeutic applications customized for ASD and other neurological disorders with genetic origins. The terms of this arrangement have been reviewed and approved by the University of California, San Diego in accordance with its conflict-of-interest policies.
Figures
References
-
- Morris CA, Mervis CB, Paciorkowski AP, Abdul-Rahman O, Dugan SL, Rope AF, Bader P, Hendon LG, Velleman SL, Klein-Tasman BP, and Osborne LR (2015). 7q11.23 Duplication syndrome: Physical characteristics and natural history. Am. J. Med. Genet 167A, 2916–2935. 10.1002/ajmg.a.37340. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R01 MH127077/MH/NIMH NIH HHS/United States
- DP2 OD006495/OD/NIH HHS/United States
- R01 DA056908/DA/NIDA NIH HHS/United States
- R01 MH123828/MH/NIMH NIH HHS/United States
- S10 OD026929/OD/NIH HHS/United States
- R01 ES033636/ES/NIEHS NIH HHS/United States
- R56 MH109587/MH/NIMH NIH HHS/United States
- U19 MH107367/MH/NIMH NIH HHS/United States
- R21 MH128827/MH/NIMH NIH HHS/United States
- T32 GM007198/GM/NIGMS NIH HHS/United States
- R01 NS105969/NS/NINDS NIH HHS/United States
- R01 HD107788/HD/NICHD NIH HHS/United States
- R01 MH100175/MH/NIMH NIH HHS/United States
- R01 HG012351/HG/NHGRI NIH HHS/United States
- R01 NS123642/NS/NINDS NIH HHS/United States
- R01 EY031318/EY/NEI NIH HHS/United States
- R01 AG078959/AG/NIA NIH HHS/United States
- R21 HD109616/HD/NICHD NIH HHS/United States
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
