

Tom20 gates PINK1 activity and mediates its tethering of the TOM and TIM23 translocases upon mitochondrial stress
- PMID: 38416681
- PMCID: PMC10927582
- DOI: 10.1073/pnas.2313540121
Tom20 gates PINK1 activity and mediates its tethering of the TOM and TIM23 translocases upon mitochondrial stress
Abstract
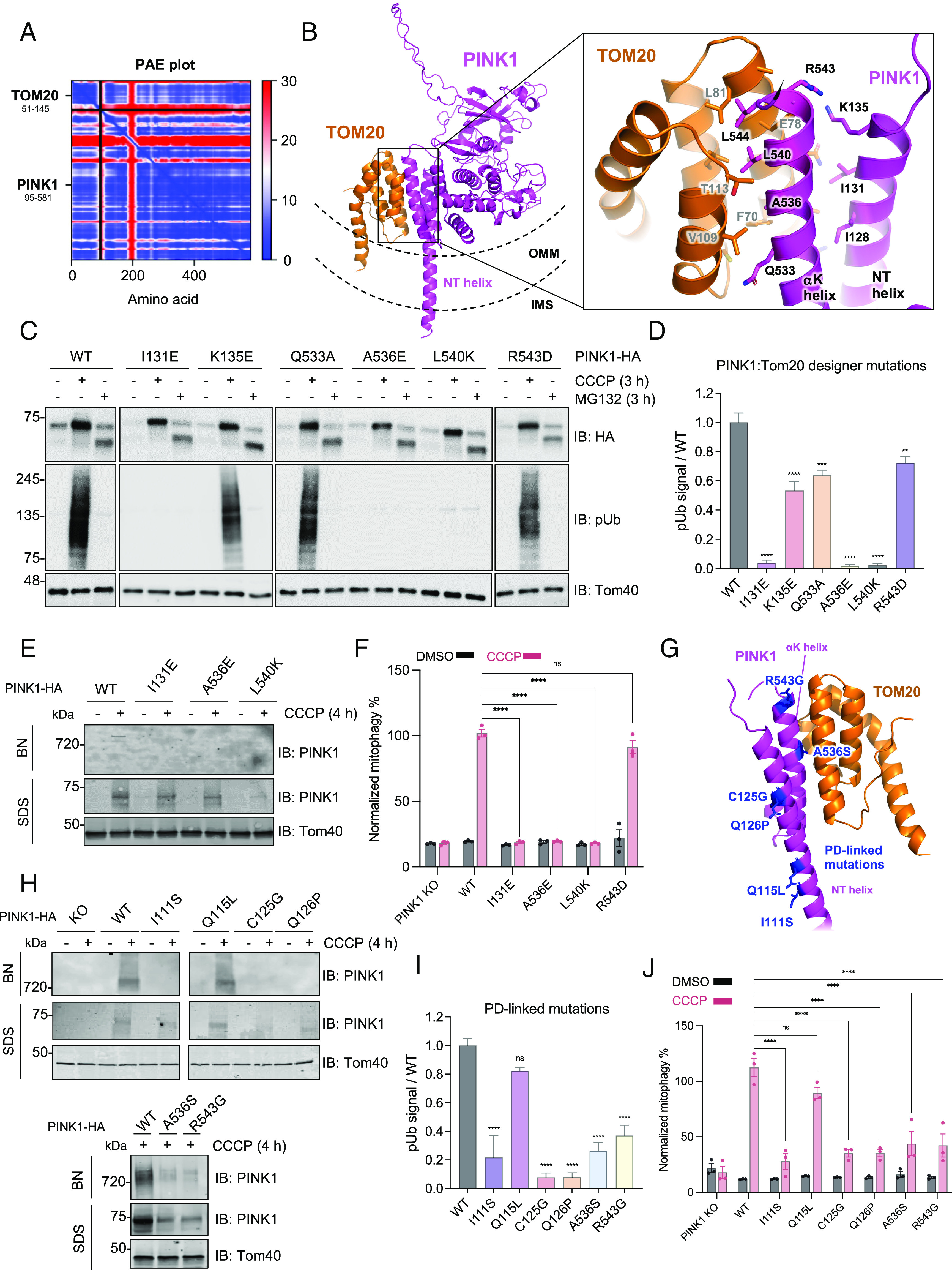
Mutations in PTEN-induced putative kinase 1 (PINK1) cause autosomal recessive early-onset Parkinson's disease (PD). PINK1 is a Ser/Thr kinase that regulates mitochondrial quality control by triggering mitophagy mediated by the ubiquitin (Ub) ligase Parkin. Upon mitochondrial damage, PINK1 accumulates on the outer mitochondrial membrane forming a high-molecular-weight complex with the translocase of the outer membrane (TOM). PINK1 then phosphorylates Ub, which enables recruitment and activation of Parkin followed by autophagic clearance of the damaged mitochondrion. Thus, Parkin-dependent mitophagy hinges on the stable accumulation of PINK1 on the TOM complex. Yet, the mechanism linking mitochondrial stressors to PINK1 accumulation and whether the translocases of the inner membrane (TIMs) are also involved remain unclear. Herein, we demonstrate that mitochondrial stress induces the formation of a PINK1-TOM-TIM23 supercomplex in human cultured cell lines, dopamine neurons, and midbrain organoids. Moreover, we show that PINK1 is required to stably tether the TOM to TIM23 complexes in response to stress such that the supercomplex fails to accumulate in cells lacking PINK1. This tethering is dependent on an interaction between the PINK1 N-terminal-C-terminal extension module and the cytosolic domain of the Tom20 subunit of the TOM complex, the disruption of which, by either designer or PD-associated PINK1 mutations, inhibits downstream mitophagy. Together, the findings provide key insight into how PINK1 interfaces with the mitochondrial import machinery, with important implications for the mechanisms of mitochondrial quality control and PD pathogenesis.
Keywords: PINK1; mitochondrial import; mitochondrial quality control; mitophagy; proteolysis.
Conflict of interest statement
Competing interests statement:J.-F.T. was a member of the scientific board of Mitokinin Inc. at the time of submission. When Mitokinin Inc. was later acquired, he received financial compensation for this consultancy work and is no longer connected with the company.
Figures



References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
