

Genome-wide enhancer-associated tandem repeats are expanded in cardiomyopathy
- PMID: 38418263
- PMCID: PMC10944212
- DOI: 10.1016/j.ebiom.2024.105027
Genome-wide enhancer-associated tandem repeats are expanded in cardiomyopathy
Abstract
Background: Cardiomyopathy is a clinically and genetically heterogeneous heart condition that can lead to heart failure and sudden cardiac death in childhood. While it has a strong genetic basis, the genetic aetiology for over 50% of cardiomyopathy cases remains unknown.
Methods: In this study, we analyse the characteristics of tandem repeats from genome sequence data of unrelated individuals diagnosed with cardiomyopathy from Canada and the United Kingdom (n = 1216) and compare them to those found in the general population. We perform burden analysis to identify genomic and epigenomic features that are impacted by rare tandem repeat expansions (TREs), and enrichment analysis to identify functional pathways that are involved in the TRE-associated genes in cardiomyopathy. We use Oxford Nanopore targeted long-read sequencing to validate repeat size and methylation status of one of the most recurrent TREs. We also compare the TRE-associated genes to those that are dysregulated in the heart tissues of individuals with cardiomyopathy.
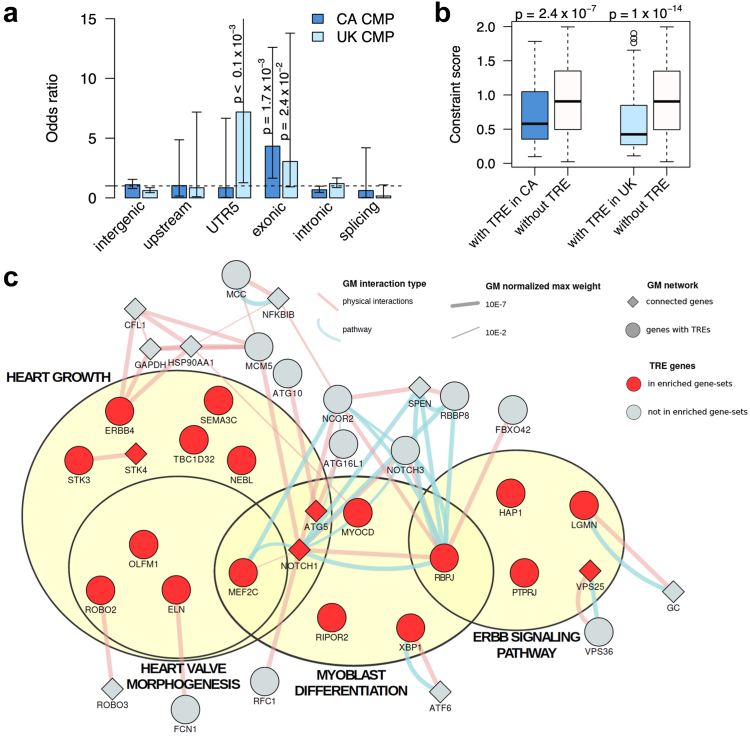
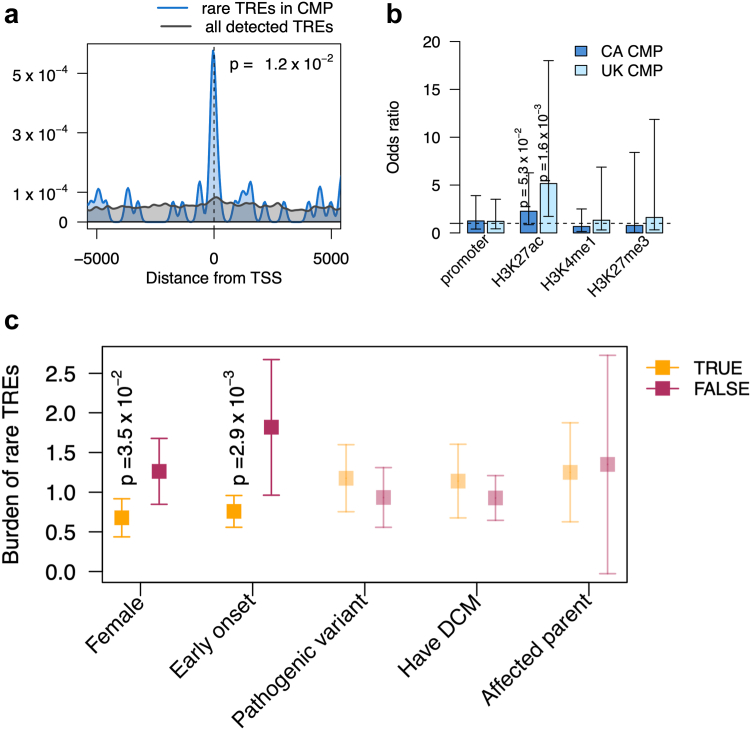
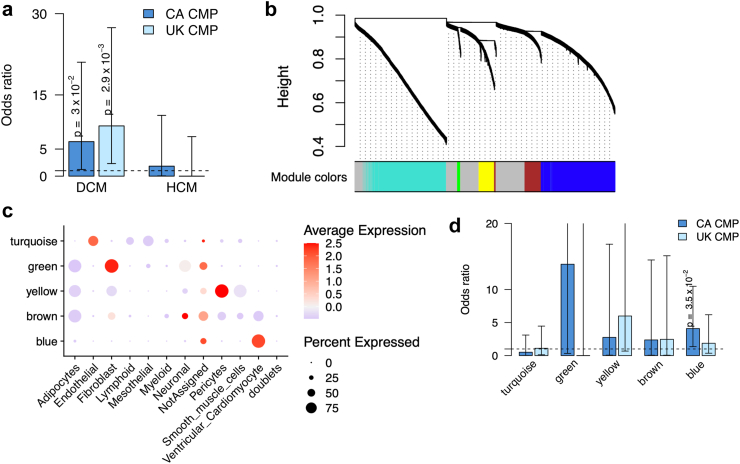
Findings: We demonstrate that tandem repeats that are rarely expanded in the general population are predominantly expanded in cardiomyopathy. We find that rare TREs are disproportionately present in constrained genes near transcriptional start sites, have high GC content, and frequently overlap active enhancer H3K27ac marks, where expansion-related DNA methylation may reduce gene expression. We demonstrate the gene silencing effect of expanded CGG tandem repeats in DIP2B through promoter hypermethylation. We show that the enhancer-associated loci are found in genes that are highly expressed in human cardiomyocytes and are differentially expressed in the left ventricle of the heart in individuals with cardiomyopathy.
Interpretation: Our findings highlight the underrecognized contribution of rare tandem repeat expansions to the risk of cardiomyopathy and suggest that rare TREs contribute to ∼4% of cardiomyopathy risk.
Funding: Government of Ontario (RKCY), The Canadian Institutes of Health Research PJT 175329 (RKCY), The Azrieli Foundation (RKCY), SickKids Catalyst Scholar in Genetics (RKCY), The University of Toronto McLaughlin Centre (RKCY, SM), Ted Rogers Centre for Heart Research (SM), Data Sciences Institute at the University of Toronto (SM), The Canadian Institutes of Health Research PJT 175034 (SM), The Canadian Institutes of Health Research ENP 161429 under the frame of ERA PerMed (SM, RL), Heart and Stroke Foundation of Ontario & Robert M Freedom Chair in Cardiovascular Science (SM), Bitove Family Professorship of Adult Congenital Heart Disease (EO), Canada Foundation for Innovation (SWS, JR), Canada Research Chair (PS), Genome Canada (PS, JR), The Canadian Institutes of Health Research (PS).
Keywords: Cardiomyopathy (CMP); Long-read sequencing; Tandem repeat expansions (TREs); Transcription regulation.
Copyright © 2024 The Author(s). Published by Elsevier B.V. All rights reserved.
Conflict of interest statement
Declaration of interests SM holds the Heart and Stroke Foundation of Canada/Robert M Freedom Chair in Cardiovascular Science and serves on the Advisory Board for Bristol Myers Squibb and Tenaya Therapeutics. EO holds the Bitove Family Professorship of Adult Congenital Heart Disease. SWS is on the Scientific Advisory Committees of Population Bio, Inc. and Deep Genomics and serves as a Highly Cited Academic Advisor for the King Abdulaziz University. SWS and RY hold patent entitled “Genome-wide Detection of DNA Repeats Expanded in Disease” (WO2021119840A1). PM is a Senior Medical Director at Maternal Newborn Child Youth Strategic Clinical Network (Alberta Health Services) and was supported for attending Clinical Microbiology and Infectious Diseases, Long COVID and ENRICH meetings. TJM received support from CIHR, PSI and SickKids Foundation, CAMH and GSK. ES received support from COVID-19 Immune Task Force, Children's Hospital Foundation and CIHR, and is on the American Academy of Allergy, Asthma, and Immunology Credentials Committee. JR is supported by Canada Foundation for Innovation and received support for attending Ultima Biosciences and 10x Genomics meetings. Illumina, Inc. provided sequencing reagents for the CHILD cohort study. CHILD cohort study was supported by CIHR, AllerGen NCE, Alberta Health, Public Health Agency of Canada and Women's and Children's Health Research. Other authors declare that they have no competing interests.
Figures


References
-
- McKenna W.J., Maron B.J., Thiene G. Classification, epidemiology, and global burden of cardiomyopathies. Circ Res. 2017;121(7):722–730. - PubMed
-
- Jensen M.K., Bundgaard H. Cardiomyopathy in Friedreich ataxia: exemplifying the challenges faced by cardiologists in the management of rare diseases. Circulation. 2012;125(13):1591–1593. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
