

A unified view on enzyme catalysis by cryo-EM study of a DNA topoisomerase
- PMID: 38418525
- PMCID: PMC10901890
- DOI: 10.1038/s42004-024-01129-y
A unified view on enzyme catalysis by cryo-EM study of a DNA topoisomerase
Abstract
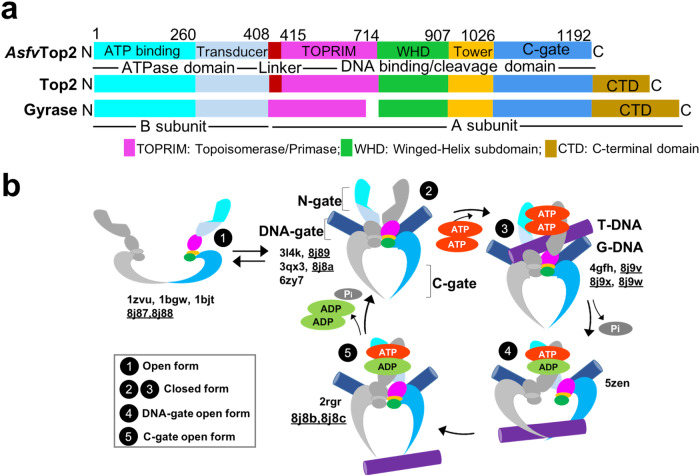
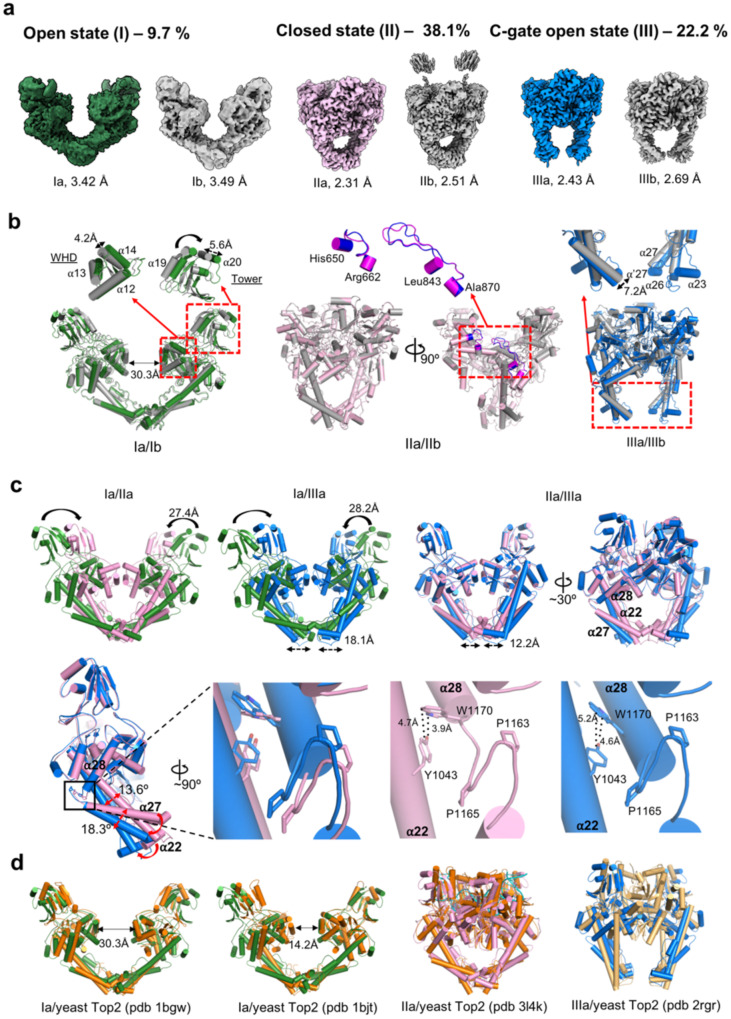
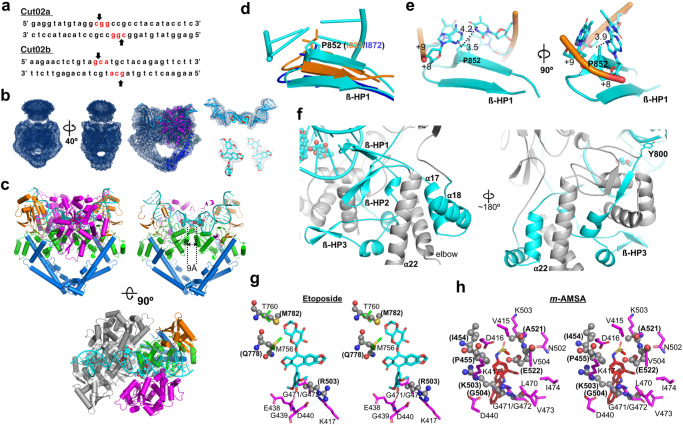
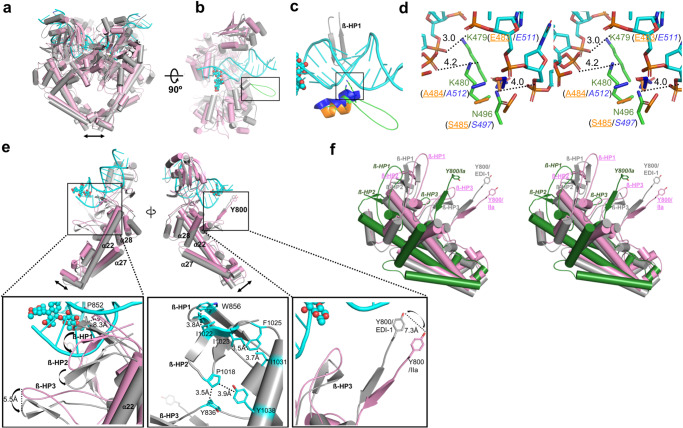
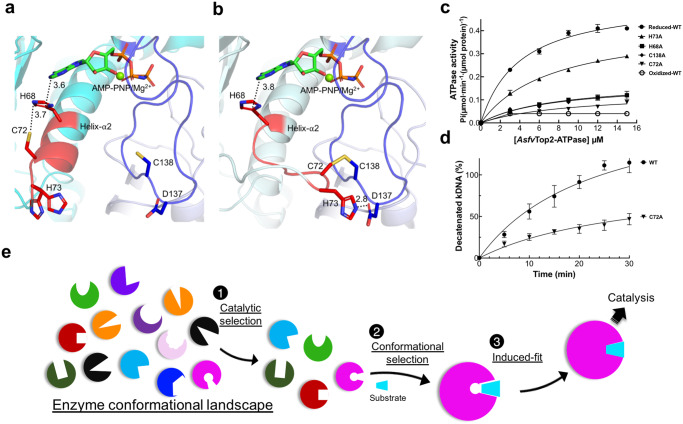
The theories for substrate recognition in enzyme catalysis have evolved from lock-key to induced fit, then conformational selection, and conformational selection followed by induced fit. However, the prevalence and consensus of these theories require further examination. Here we use cryogenic electron microscopy and African swine fever virus type 2 topoisomerase (AsfvTop2) to demonstrate substrate binding theories in a joint and ordered manner: catalytic selection by the enzyme, conformational selection by the substrates, then induced fit. The apo-AsfvTop2 pre-exists in six conformers that comply with the two-gate mechanism directing DNA passage and release in the Top2 catalytic cycle. The structures of AsfvTop2-DNA-inhibitor complexes show that substantial induced-fit changes occur locally from the closed apo-conformer that however is too far-fetched for the open apo-conformer. Furthermore, the ATPase domain of AsfvTop2 in the MgAMP-PNP-bound crystal structures coexist in reduced and oxidized forms involving a disulfide bond, which can regulate the AsfvTop2 function.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Fischer E. Einfluss der configuration auf die wirkung der enzyme. Ber. Dtsch. Chem. Ges. 1894;27:2985–1993. doi: 10.1002/cber.18940270364. - DOI
LinkOut - more resources
Full Text Sources
Miscellaneous
