

Durable and efficient gene silencing in vivo by hit-and-run epigenome editing
- PMID: 38418872
- PMCID: PMC10937395
- DOI: 10.1038/s41586-024-07087-8
Durable and efficient gene silencing in vivo by hit-and-run epigenome editing
Abstract
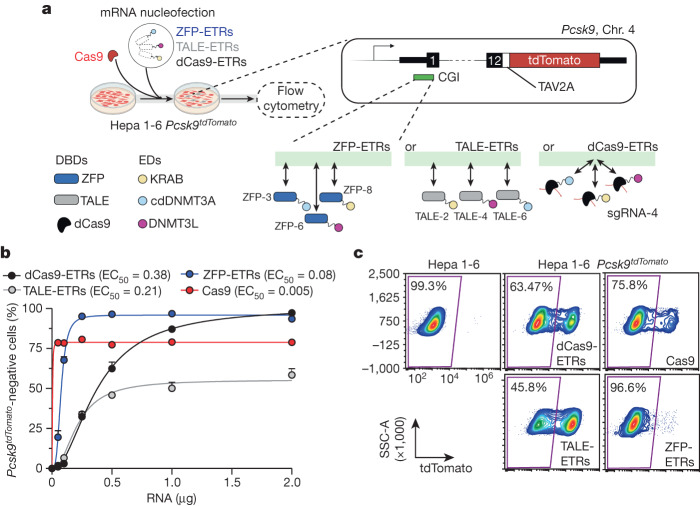
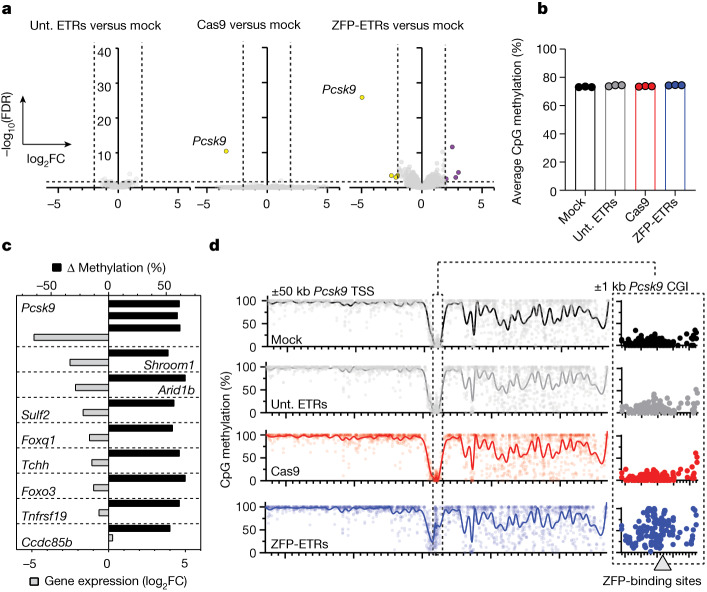
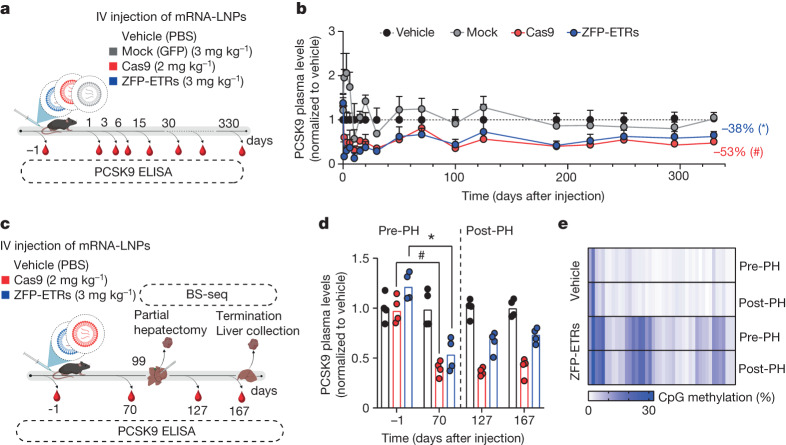
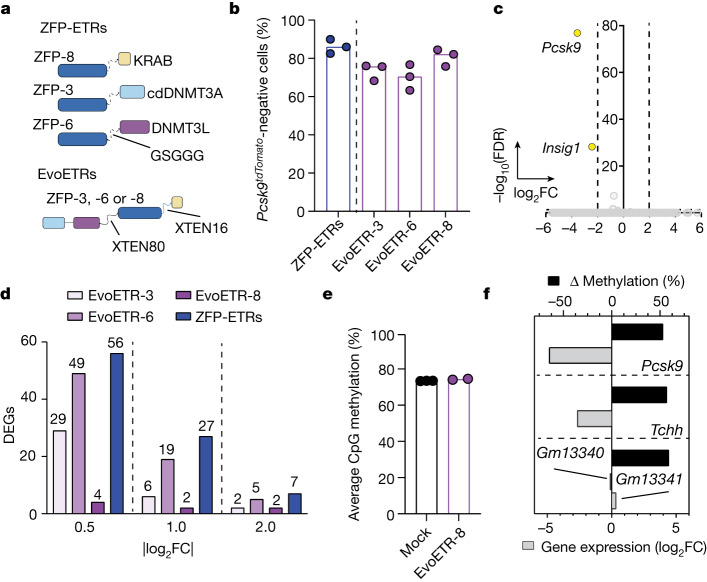
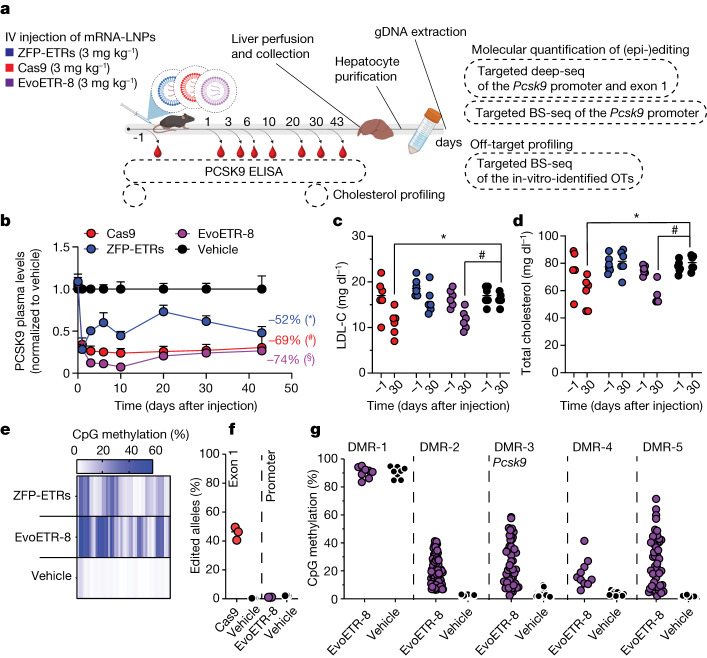
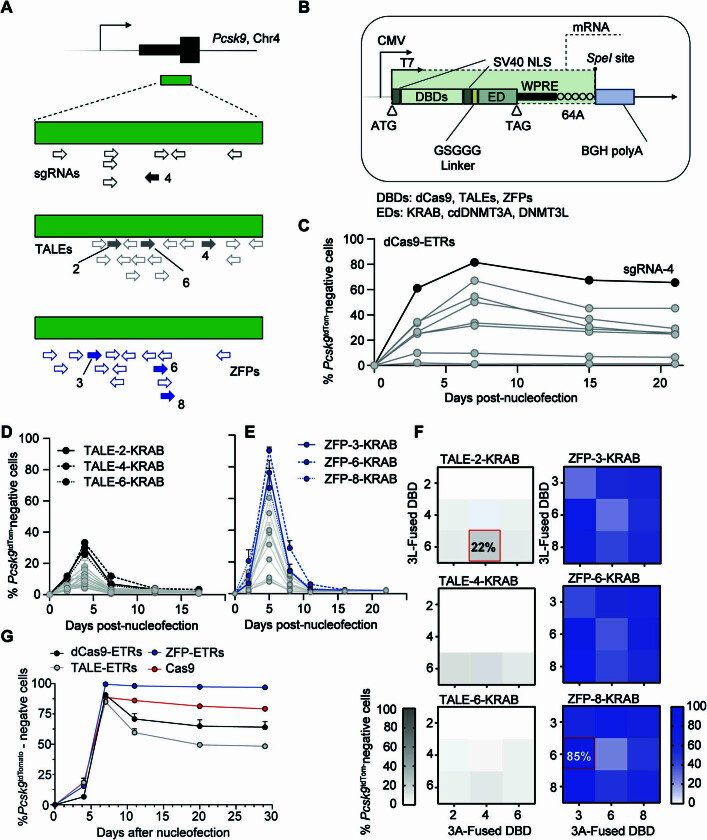
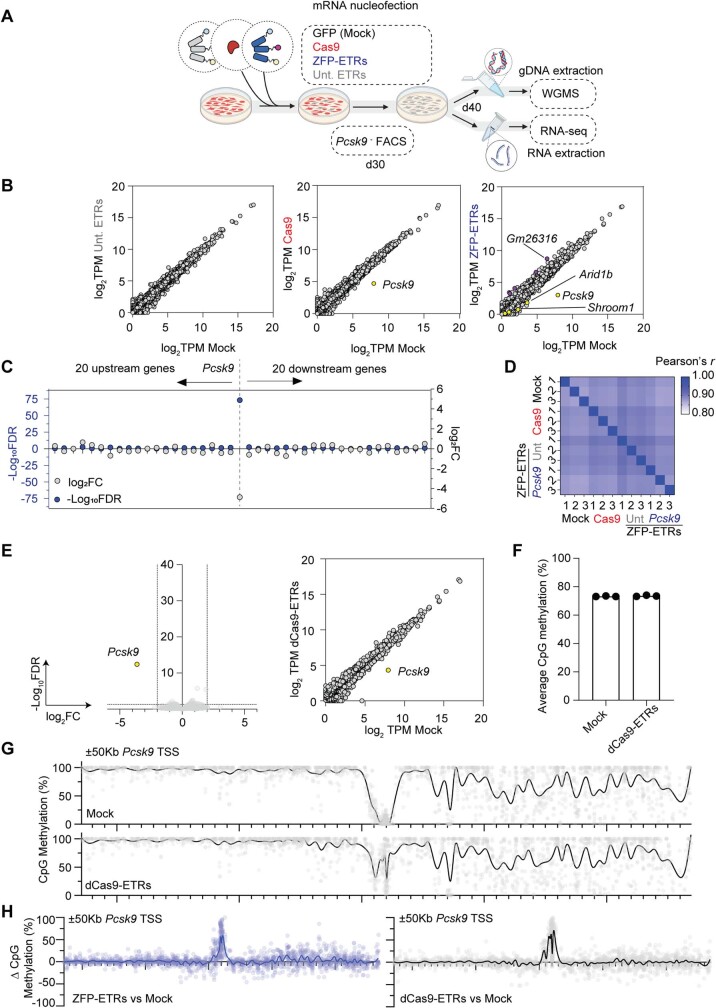
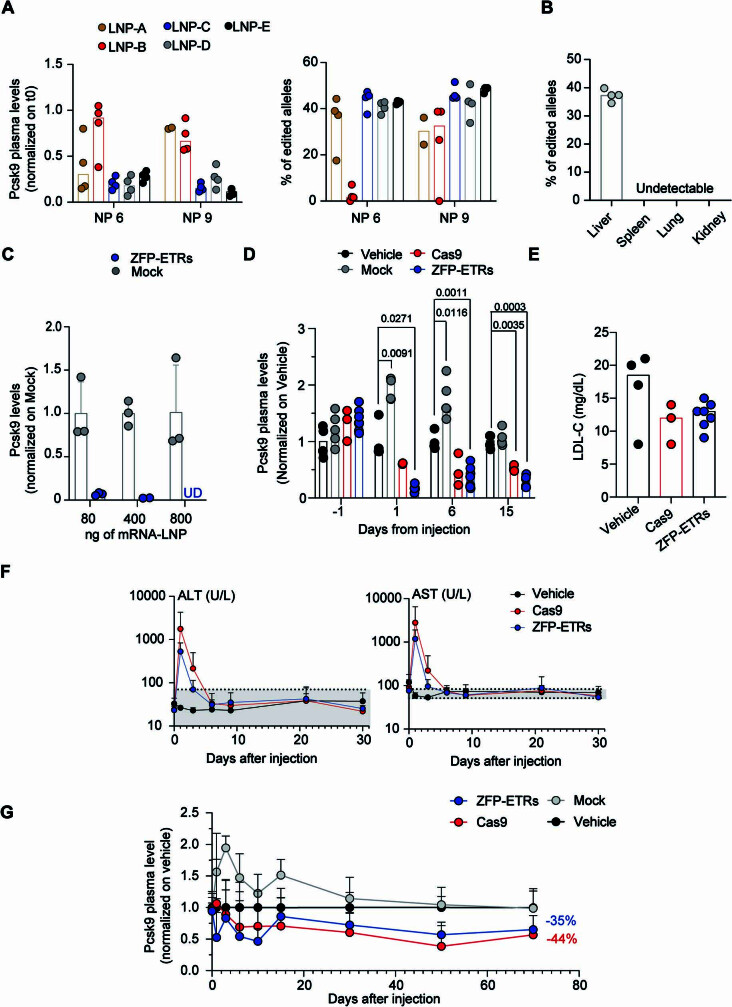
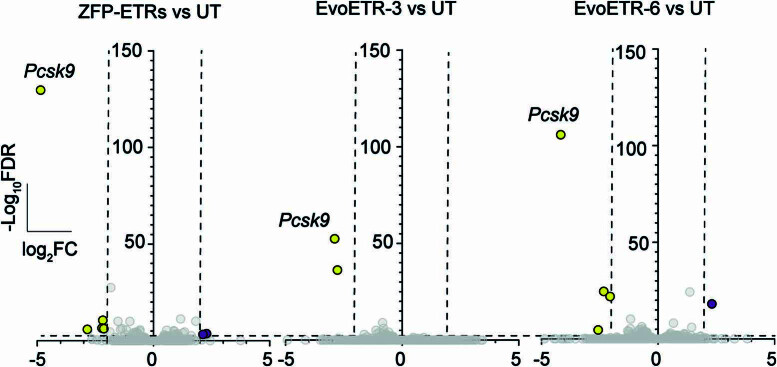
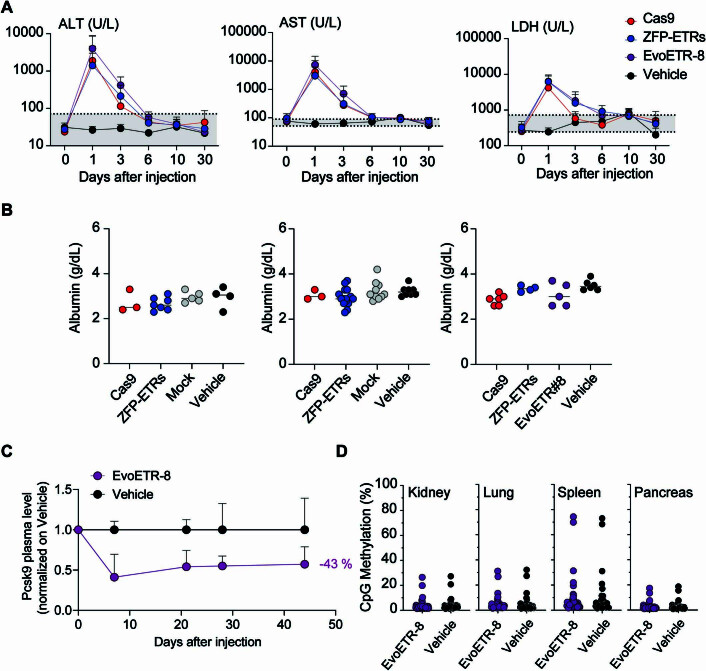
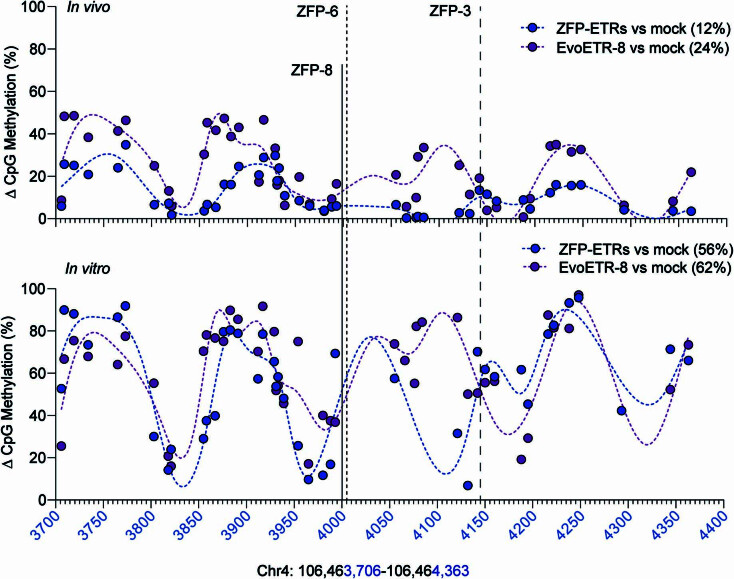
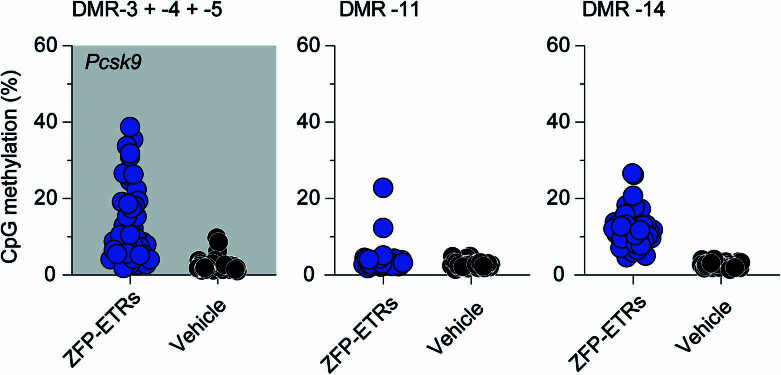
Permanent epigenetic silencing using programmable editors equipped with transcriptional repressors holds great promise for the treatment of human diseases1-3. However, to unlock its full therapeutic potential, an experimental confirmation of durable epigenetic silencing after the delivery of transient delivery of editors in vivo is needed. To this end, here we targeted Pcsk9, a gene expressed in hepatocytes that is involved in cholesterol homeostasis. In vitro screening of different editor designs indicated that zinc-finger proteins were the best-performing DNA-binding platform for efficient silencing of mouse Pcsk9. A single administration of lipid nanoparticles loaded with the editors' mRNAs almost halved the circulating levels of PCSK9 for nearly one year in mice. Notably, Pcsk9 silencing and accompanying epigenetic repressive marks also persisted after forced liver regeneration, further corroborating the heritability of the newly installed epigenetic state. Improvements in construct design resulted in the development of an all-in-one configuration that we term evolved engineered transcriptional repressor (EvoETR). This design, which is characterized by a high specificity profile, further reduced the circulating levels of PCSK9 in mice with an efficiency comparable with that obtained through conventional gene editing, but without causing DNA breaks. Our study lays the foundation for the development of in vivo therapeutics that are based on epigenetic silencing.
© 2024. The Author(s).
Conflict of interest statement
A.L. is a co-founder of, quota holder of and consultant for Chroma Medicine, a company aiming to develop epi-editing applications. A.L. and M.A.C. are inventors on pending and issued patents related to epi-silencing filed by the San Raffaele Scientific Institute and Telethon Foundation, or Chroma Medicine. The remaining authors declare no competing interests.
Figures
Comment in
-
An epigenome editing approach induces durable silencing of Pcsk9.Nat Rev Cardiol. 2024 May;21(5):279. doi: 10.1038/s41569-024-01011-3. Nat Rev Cardiol. 2024. PMID: 38480792 No abstract available.
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
