

Anti-TIGIT antibody improves PD-L1 blockade through myeloid and Treg cells
- PMID: 38418879
- PMCID: PMC11139643
- DOI: 10.1038/s41586-024-07121-9
Anti-TIGIT antibody improves PD-L1 blockade through myeloid and Treg cells
Erratum in
-
Publisher Correction: Anti-TIGIT antibody improves PD-L1 blockade through myeloid and Treg cells.Nature. 2024 Mar;627(8005):E11. doi: 10.1038/s41586-024-07280-9. Nature. 2024. PMID: 38480897 No abstract available.
-
Publisher Correction: Anti-TIGIT antibody improves PD-L1 blockade through myeloid and Treg cells.Nature. 2024 Jun;630(8016):E9. doi: 10.1038/s41586-024-07562-2. Nature. 2024. PMID: 38816613 Free PMC article. No abstract available.
-
Author Correction: Anti-TIGIT antibody improves PD-L1 blockade through myeloid and Treg cells.Nature. 2024 Sep;633(8030):E1. doi: 10.1038/s41586-024-07956-2. Nature. 2024. PMID: 39164390 Free PMC article. No abstract available.
Abstract
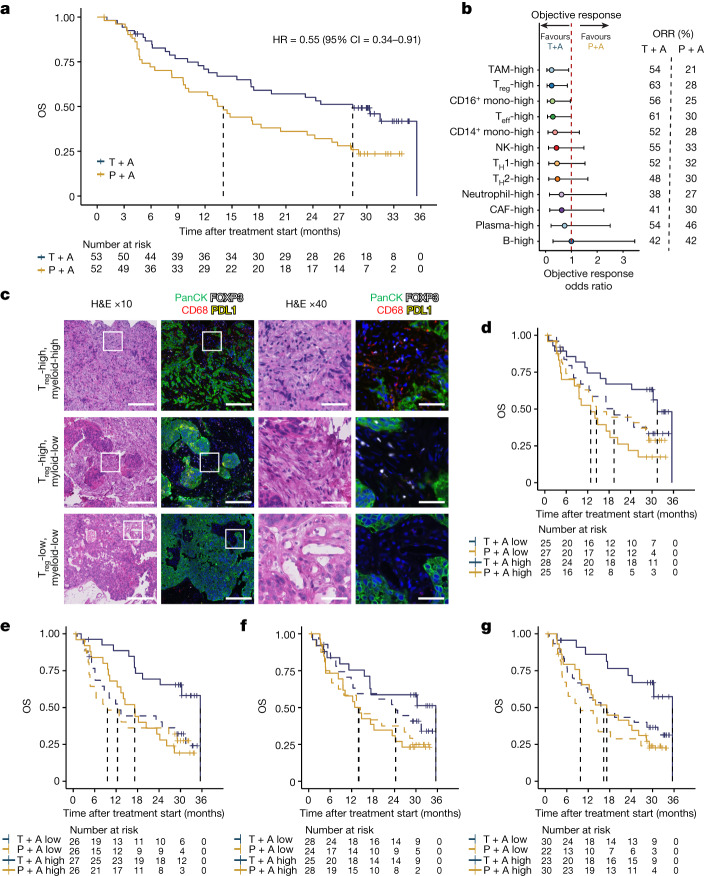
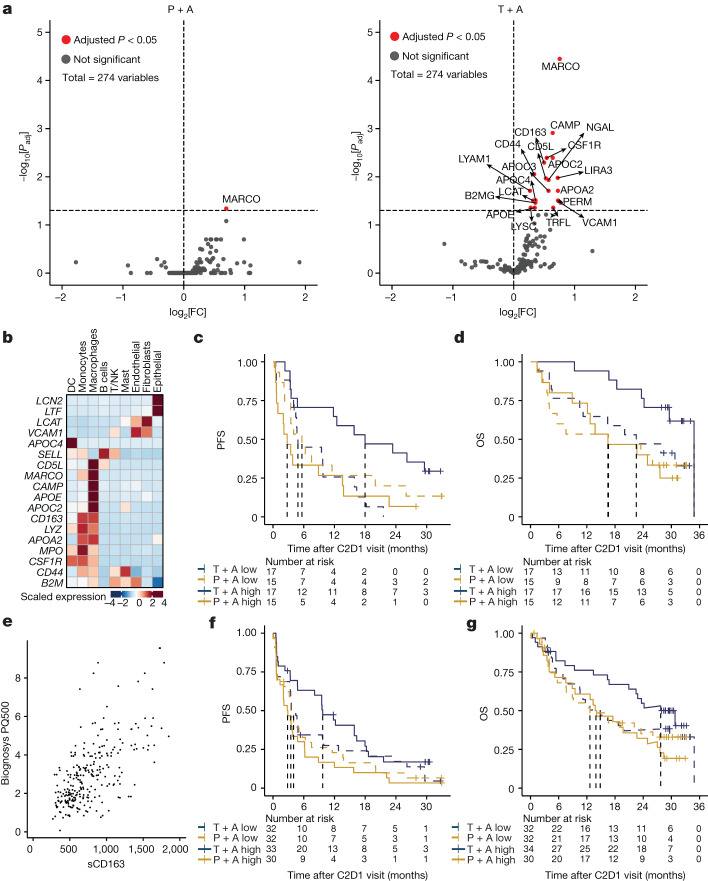
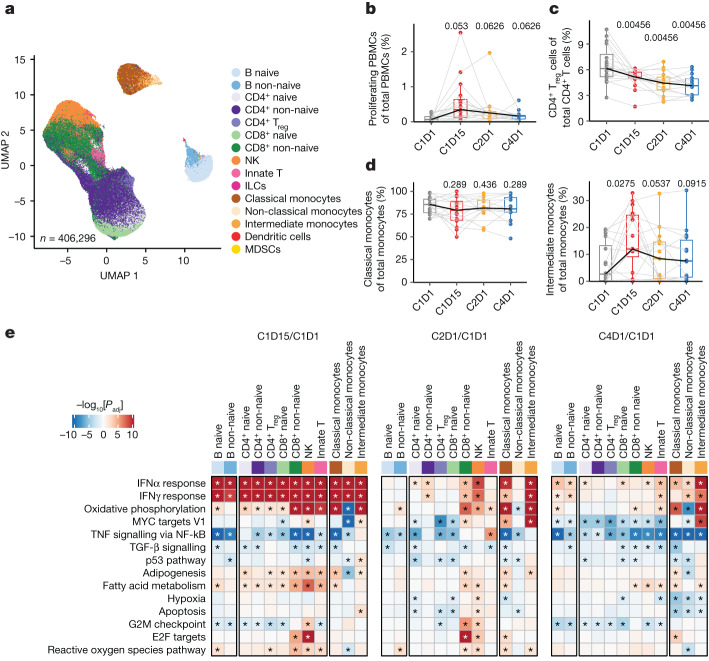
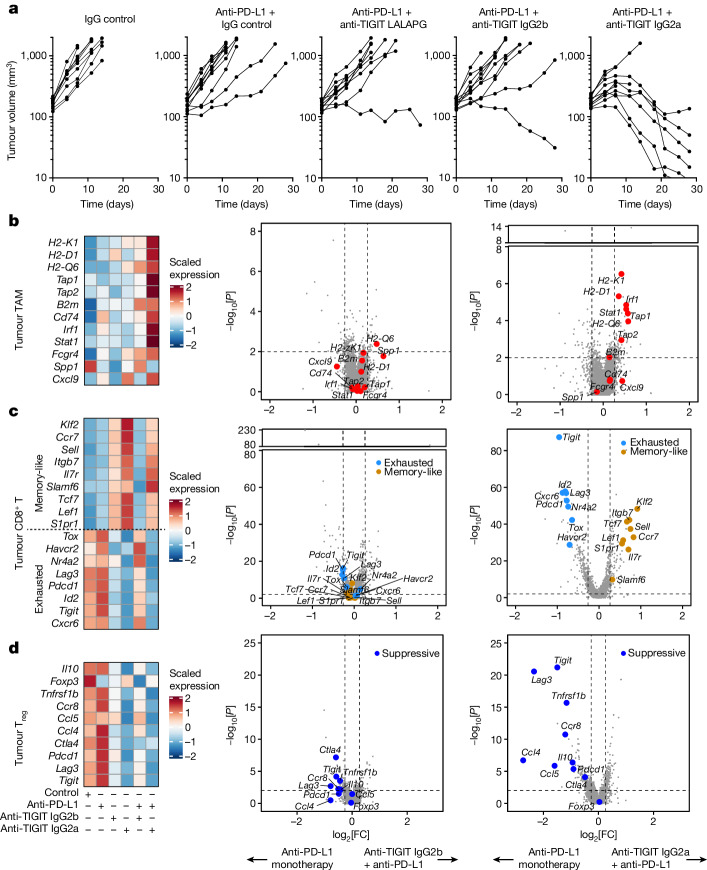
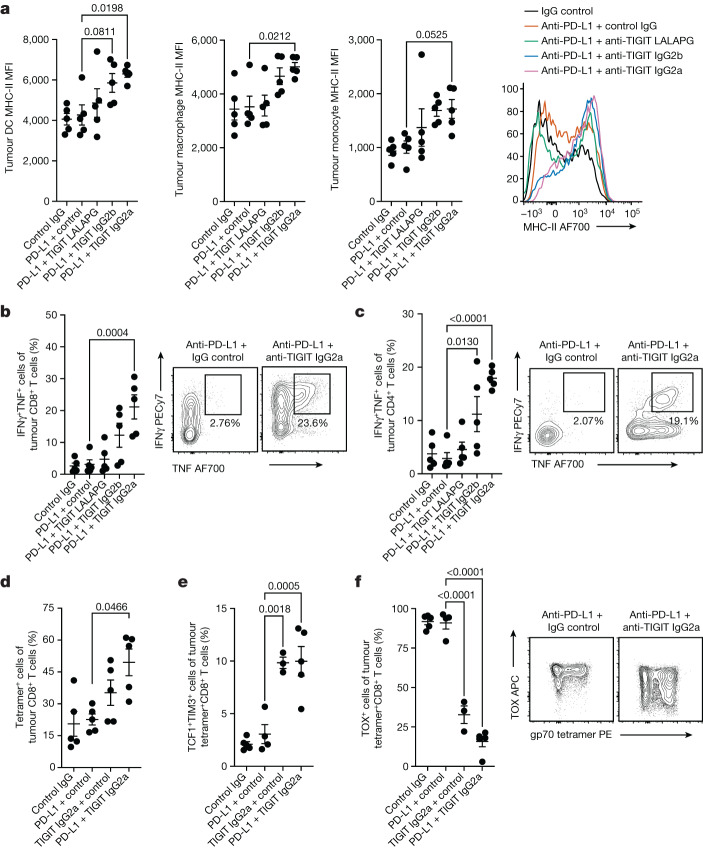
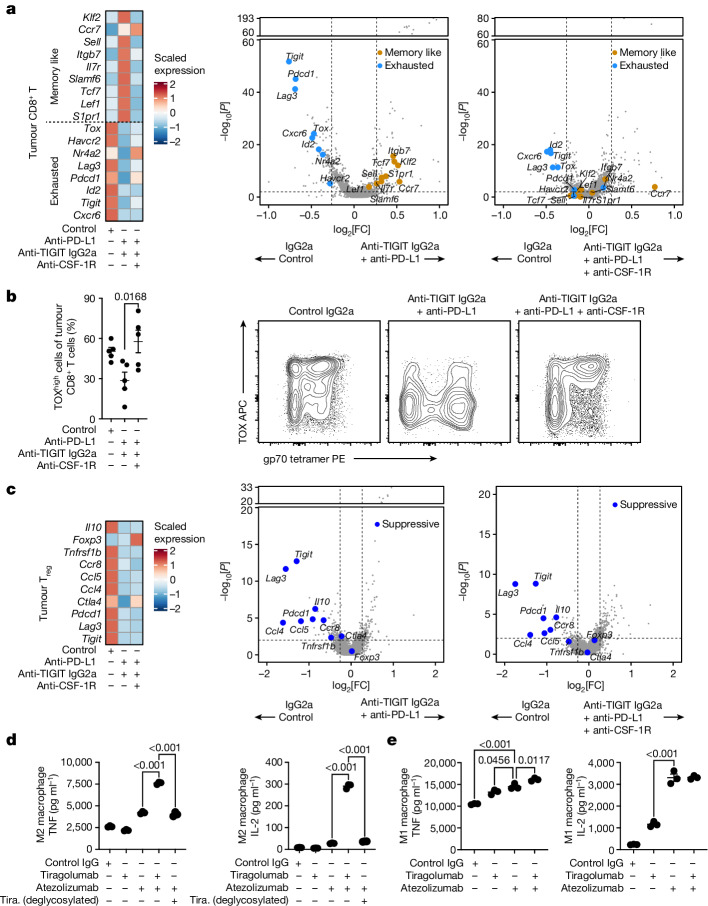
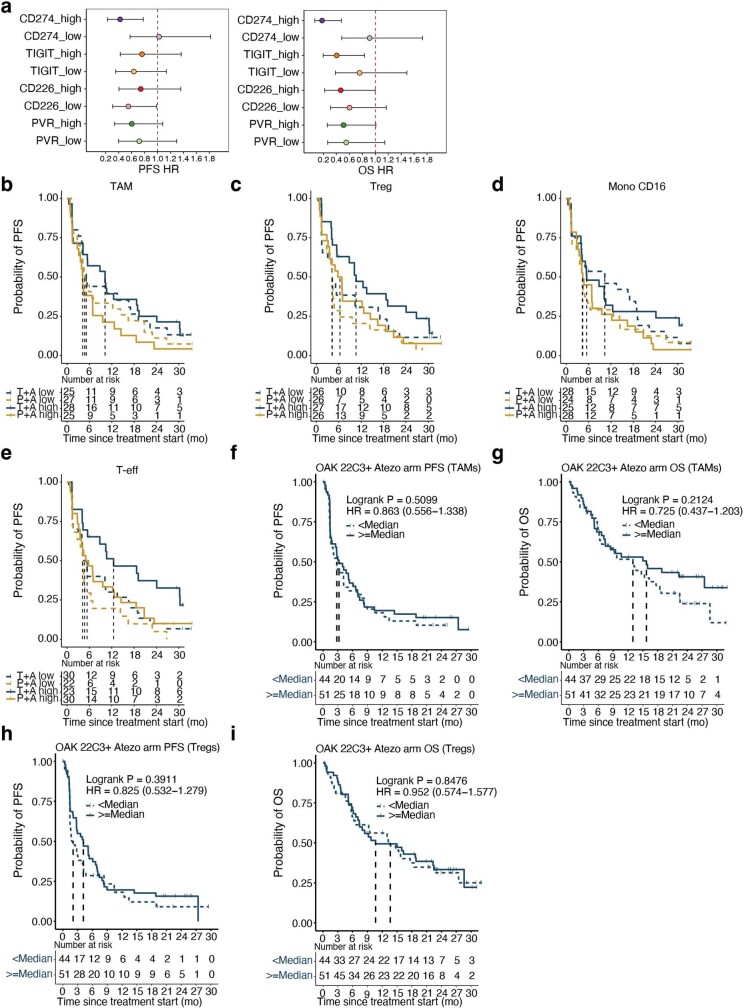
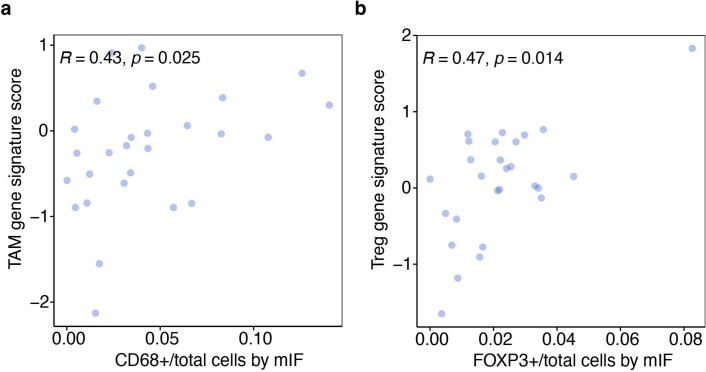
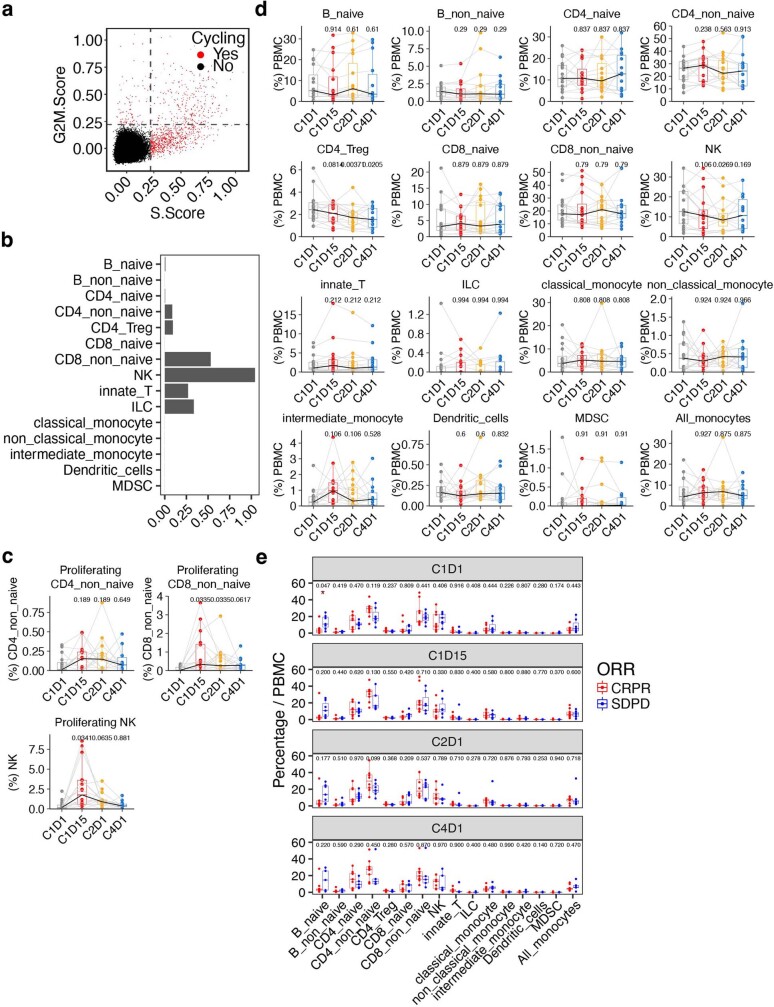
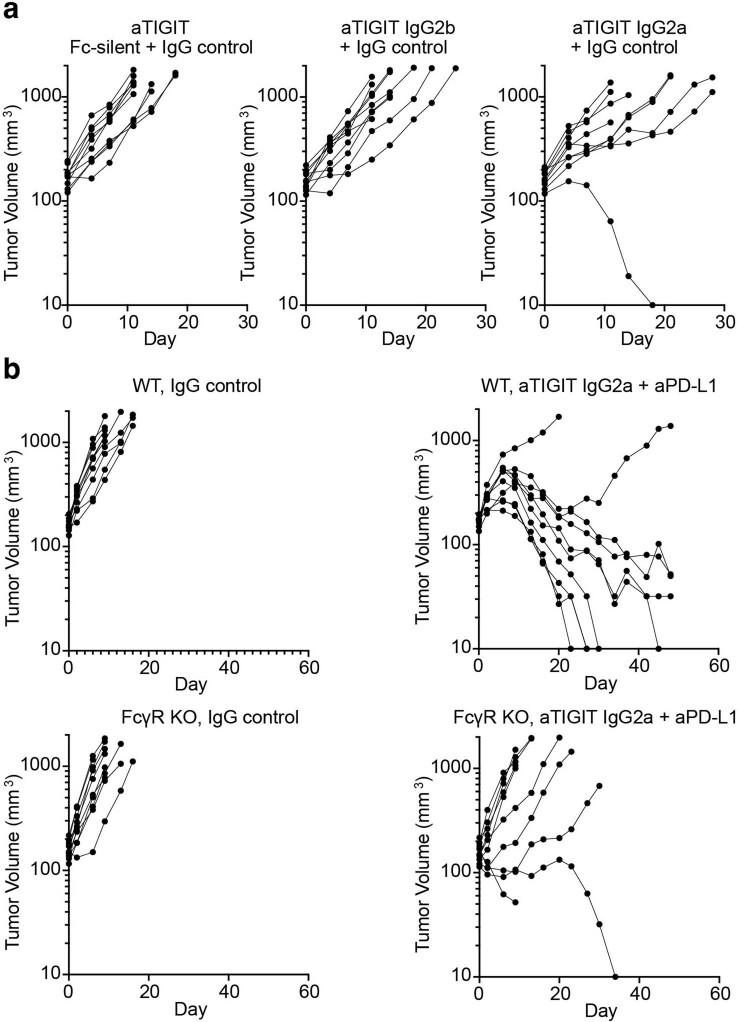
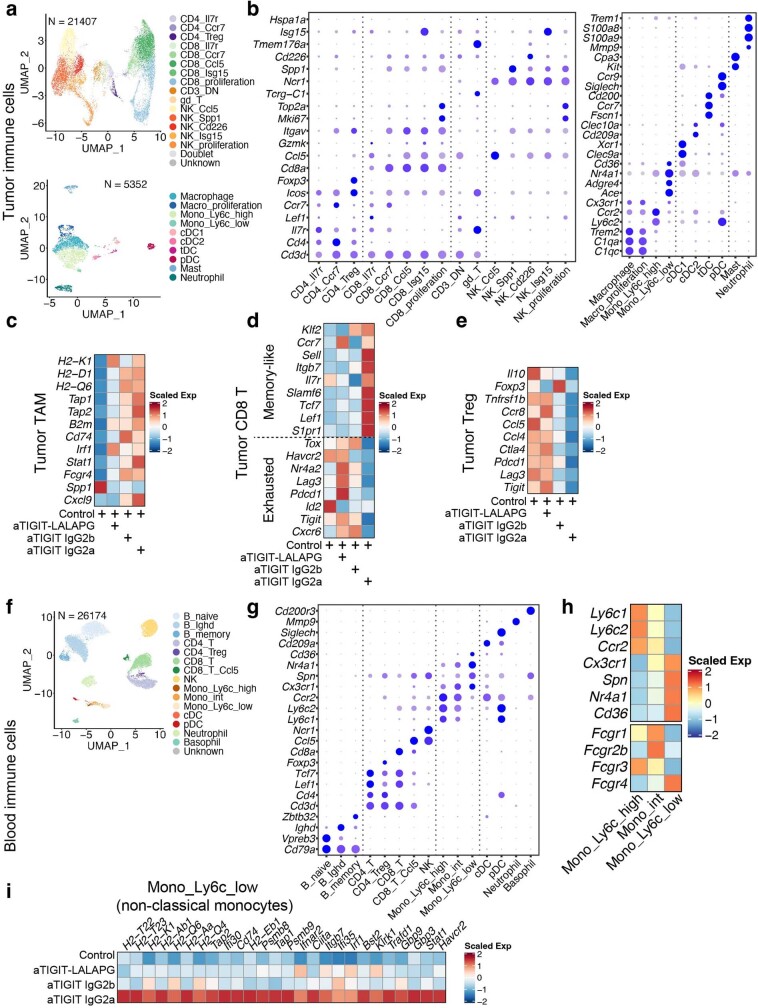
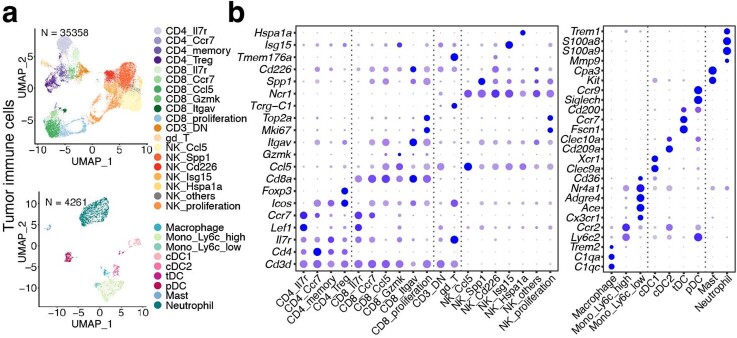
Tiragolumab, an anti-TIGIT antibody with an active IgG1κ Fc, demonstrated improved outcomes in the phase 2 CITYSCAPE trial (ClinicalTrials.gov: NCT03563716 ) when combined with atezolizumab (anti-PD-L1) versus atezolizumab alone1. However, there remains little consensus on the mechanism(s) of response with this combination2. Here we find that a high baseline of intratumoural macrophages and regulatory T cells is associated with better outcomes in patients treated with atezolizumab plus tiragolumab but not with atezolizumab alone. Serum sample analysis revealed that macrophage activation is associated with a clinical benefit in patients who received the combination treatment. In mouse tumour models, tiragolumab surrogate antibodies inflamed tumour-associated macrophages, monocytes and dendritic cells through Fcγ receptors (FcγR), in turn driving anti-tumour CD8+ T cells from an exhausted effector-like state to a more memory-like state. These results reveal a mechanism of action through which TIGIT checkpoint inhibitors can remodel immunosuppressive tumour microenvironments, and suggest that FcγR engagement is an important consideration in anti-TIGIT antibody development.
© 2024. The Author(s), under exclusive licence to Springer Nature Limited.
Conflict of interest statement
X.G., R. Hu, Y.C., S.S., B.Y.N., J.S., L.M., R. Hendricks, K.N., K.L.B., E.D., A.D., P.S.C., J.H., S. Mittman, N.M., P.D., W.C., I.M., S. Mariathasan, D.S.S., R.M., E.Y.C., R.J.J. and N.S.P. are employees and stockholders of Roche/Genentech. M.J. declares research funding (paid to institution) from AbbVie, Acerta, Adaptimmune, Amgen, Apexigen, Arcus Biosciences, Array BioPharma, ArriVent BioPharma, Artios Pharma, AstraZeneca, Atreca, BeiGene, BerGenBio, BioAtla, Black Diamond, Boehringer Ingelheim, Bristol-Myers Squibb, Calithera Biosciences, Carisma Therapeutics, Checkpoint Therapeutics, City of Hope National Medical Center, Corvus Pharmaceuticals, Curis, CytomX, Daiichi Sankyo, Dracen Pharmaceuticals, Dynavax, Lilly, Eikon Therapeutics, Elicio Therapeutics, EMD Serono, EQRx, Erasca, Exelixis, Fate Therapeutics, Genentech/Roche, Genmab, Genocea Biosciences, GlaxoSmithKline, Gritstone Oncology, Guardant Health, Harpoon, Helsinn Healthcare SA, Hengrui Therapeutics, Hutchison MediPharma, IDEAYA Biosciences, IGM Biosciences, Immunitas Therapeutics, Immunocore, Incyte, Janssen, Jounce Therapeutics, Kadmon Pharmaceuticals, Kartos Therapeutics, LockBody Therapeutics, Loxo Oncology, Lycera, Memorial Sloan-Kettering, Merck, Merus, Mirati Therapeutics, Mythic Therapeutics, NeoImmune Tech, Neovia Oncology, Novartis, Numab Therapeutics, Nuvalent, OncoMed Pharmaceuticals, Palleon Pharmaceuticals, Pfizer, PMV Pharmaceuticals, Rain Therapeutics, RasCal Therapeutics, Regeneron Pharmaceuticals, Relay Therapeutics, Revolution Medicines, Ribon Therapeutics, Rubius Therapeutics, Sanofi, Seven and Eight Biopharmaceuticals/Birdie Biopharmaceuticals, Shattuck Labs, Silicon Therapeutics, Stem CentRx, Syndax Pharmaceuticals, Taiho Oncology, Takeda Pharmaceuticals, Tarveda, TCR2 Therapeutics, Tempest Therapeutics, Tizona Therapeutics, TMUNITY Therapeutics, Turning Point Therapeutics, University of Michigan, Vyriad, WindMIL Therapeutics and Y-mAbs Therapeutics; and consulting/advisory roles (paid to institution) for AbbVie, Amgen, Arcus Biosciences, Arrivent, Astellas, AstraZeneca, Boehringer Ingelheim, Bristol-Myers Squibb, Calithera Biosciences, D3 Bio Limited, Daiichi Sankyo, Fate Therapeutics, Genentech/Roche, Genmab, Genocea Biosciences, Gilead Sciences, GlaxoSmithKline, Gritstone Oncology, Hookipa Biotech, Immunocore, Janssen, Jazz Pharmaceuticals, Lilly, Merck, Mirati Therapeutics, Molecular Axiom, Normunity, Novartis, Novocure, Pfizer, Pyramid Biosciences, Revolution Medicines, Sanofi-Aventis, SeaGen, Synthekine, Takeda Pharmaceuticals and VBL Therapeutics. D.R.A. reports personal payment/honoraria from Roche, AstraZeneca, Bristol-Myers Squibb, Merck Sharp & Dohme, Eli Lilly, Pfizer, and Novartis; and institutional support for attending meetings or travel from Roche, Bristol-Myers Squibb, Merck Sharp & Dohme and Novartis. BCC declares royalties from Champions Oncology, Crown Bioscience, Imagen and PearlRiver Bio; grants/research support/funding from MOGAM Institute, LG Chem, Oscotec, Interpark Bio Convergence Corp, GIInnovation, GI-Cell, Abion, Abbvie, AstraZeneca, Bayer, Blueprint Medicines, Boehringer Ingelheim, Champions Oncology, CJ Bioscience, CJ Blossom Park, Cyrus, Dizal Pharma, Genexine, Janssen, Lilly, MSD, Novartis, Nuvalent, Oncternal, Ono, Regeneron, Dong-A ST, Bridgebio Therapeutics, Yuhan, ImmuneOncia, Illumina, Kanaph Therapeutics, Therapex, JINTSbio, Hanmi, CHA Bundang Medical Center and Vertical Bio AG; consultancy roles for Abion, BeiGene, Novartis, AstraZeneca, Boehringer-Ingelheim, Roche, BMS, CJ, CureLogen, Cyrus Therapeutics, Ono, Onegene Biotechnology, Yuhan, Pfizer, Eli Lilly, GI-Cell, Guardant, HK Inno-N, Imnewrun Biosciences, Janssen, Takeda, MSD, Medpacto, Blueprint medicines, RandBio and Hanmi; employment from Yonsei University Health System; participation on an advisory board for KANAPH Therapeutic, Bridgebio Therapeutics, Cyrus Therapeutics, Guardant Health, Oscotec, J INTS Bio, Therapex, Gliead and Amgen; speaker roles for ASCO, AstraZeneca, Guardant, Roche, ESMO, IASLC, Korean Cancer Association, Korean Society of Medical Onoclogy, Korean Society of Thyroid-Head and Neck Surgery, Korean Cancer Study Group, Novartis, MSD, The Chinese Thoracic Oncology Society and Pfizer; stocks/shares in TheraCanVac, Gencurix, Bridgebio Therapeutics, KANAPH Therapeutic, Cyrus Therapeutics, Interpark Bio Convergence and J INTS BIO; founder for DAAN Biotherapeutics; and member of the board of directors for J INTS BIO. A.I. declares grants and/or consulting fees from BMS, MSD, Roche, Bayer and AstraZeneca. I.G.-B. declares clinical investigator, advisory board, speaker and director of scientific meeting roles for Roche/Genentech; and financial support from Roche/Genentech. E.F. declares advisory board/speaker roles for Abbvie, Amgen, Astra Zeneca, Bayer, Beigene, Boehringer Ingelheim, Bristol Myers Squibb, Daiichi Sankyo, Eli Lilly, F. Hoffmann-La Roche, Genentech, Gilead, Glaxo Smith Kline, Janssen, Medscape, Merck Serono, Merck Sharp & Dohme, Novartis, Peervoice, Peptomyc, Pfizer, Regeneron, Sanofi, Takeda, Touch Oncology and Turning Point Therapeutics; and independent member of the board for Grifols.
Figures
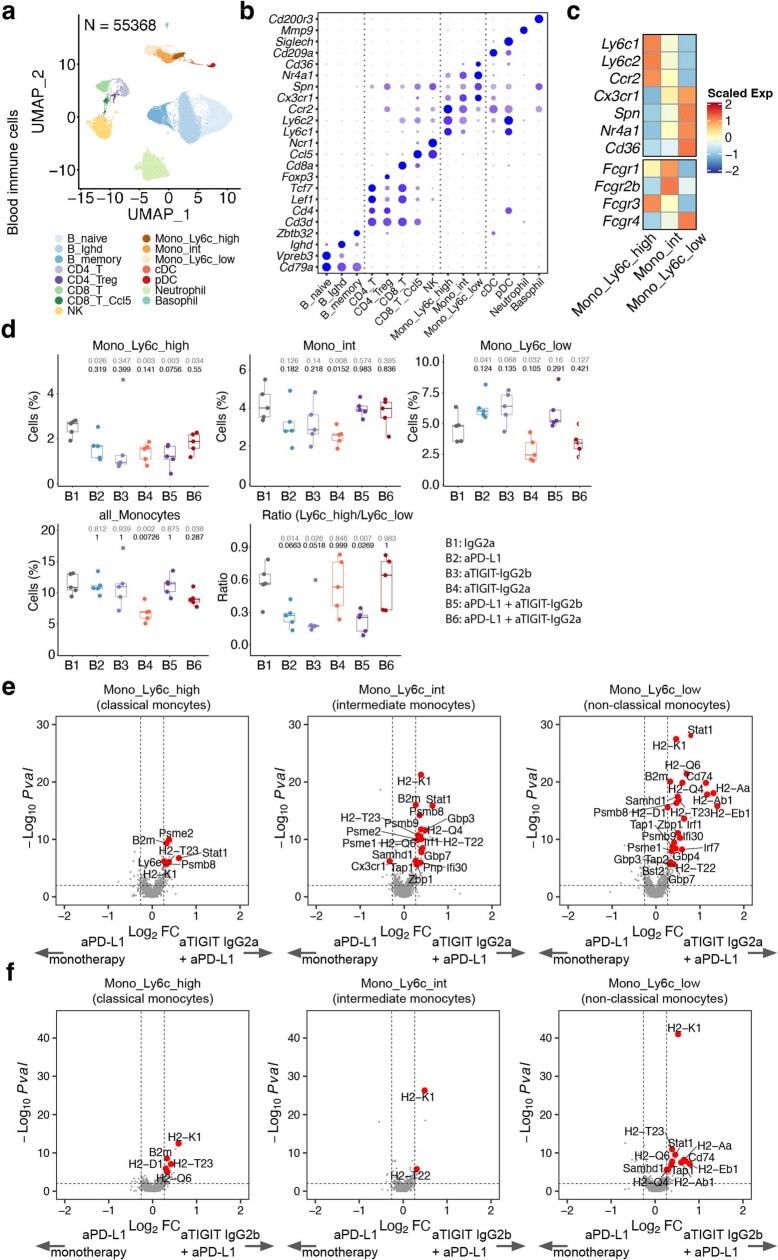
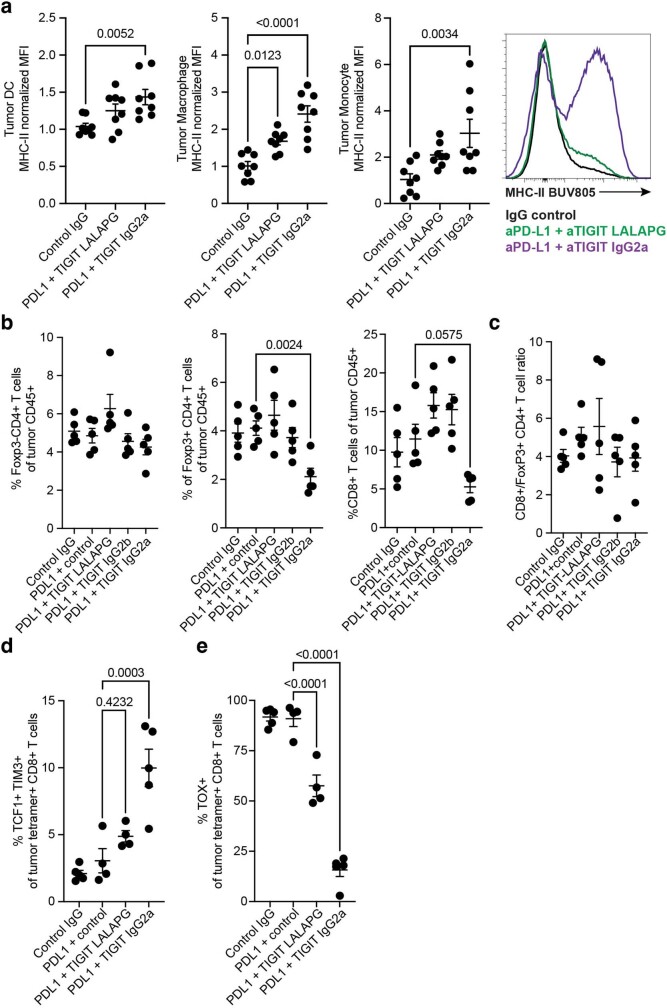
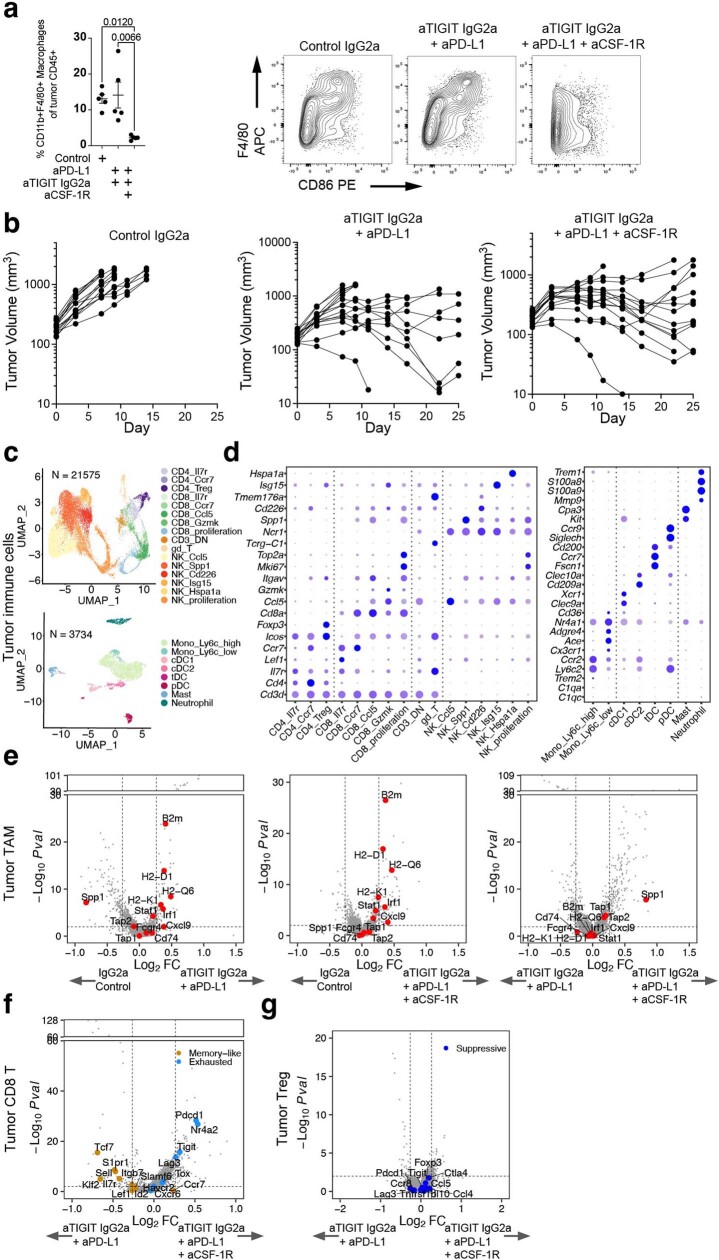
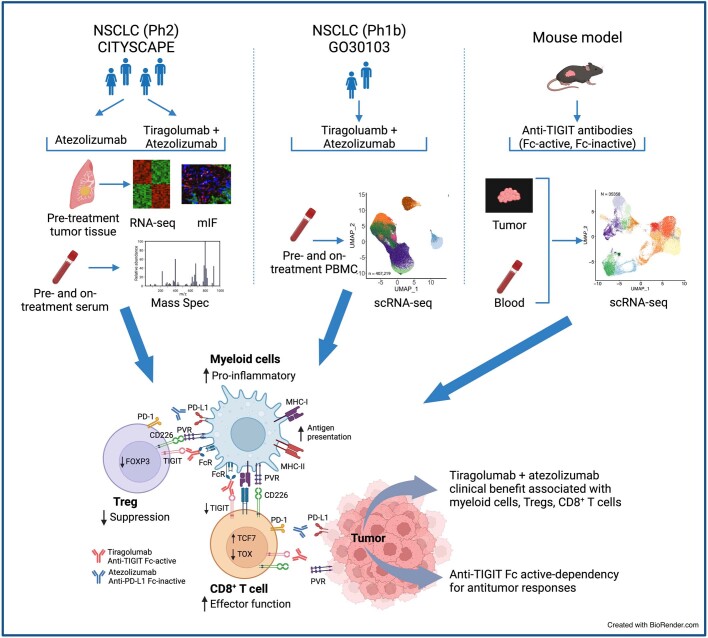
References
-
- Cho, B. C. et al. Tiragolumab plus atezolizumab versus placebo plus atezolizumab as a first-line treatment for PD-L1-selected non-small-cell lung cancer (CITYSCAPE): primary and follow-up analyses of a randomised, double-blind, phase 2 study. Lancet Oncol.23, 781–792 (2022). - PubMed
-
- Dolgin, E. Antibody engineers seek optimal drug targeting TIGIT checkpoint. Nat. Biotechnol.38, 1007–1009 (2020). - PubMed
-
- Wang, M., Herbst, R. S. & Boshoff, C. Toward personalized treatment approaches for non-small-cell lung cancer. Nat. Med.27, 1345–1356 (2021). - PubMed
MeSH terms
Substances
Associated data
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
