

YAP-mediated GPER signaling impedes proliferation and survival of prostate epithelium in benign prostatic hyperplasia
- PMID: 38420594
- PMCID: PMC10901089
- DOI: 10.1016/j.isci.2024.109125
YAP-mediated GPER signaling impedes proliferation and survival of prostate epithelium in benign prostatic hyperplasia
Abstract
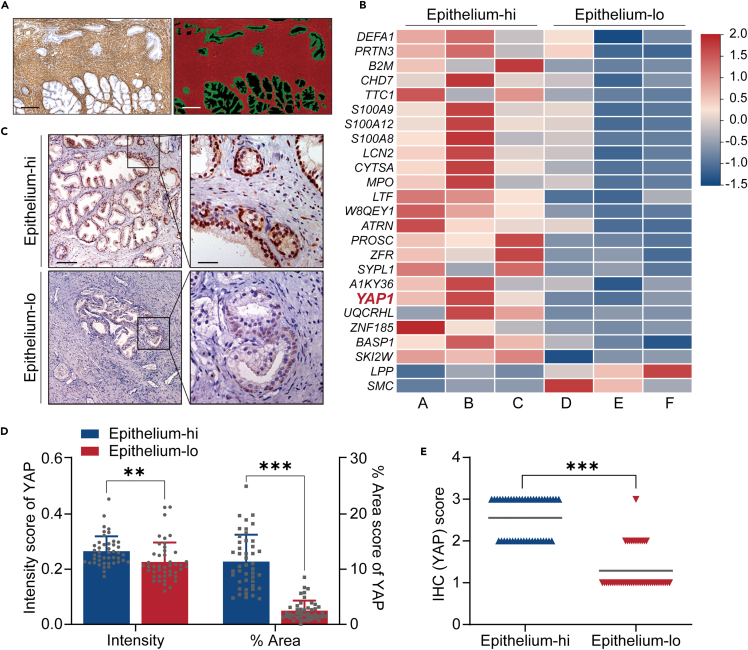
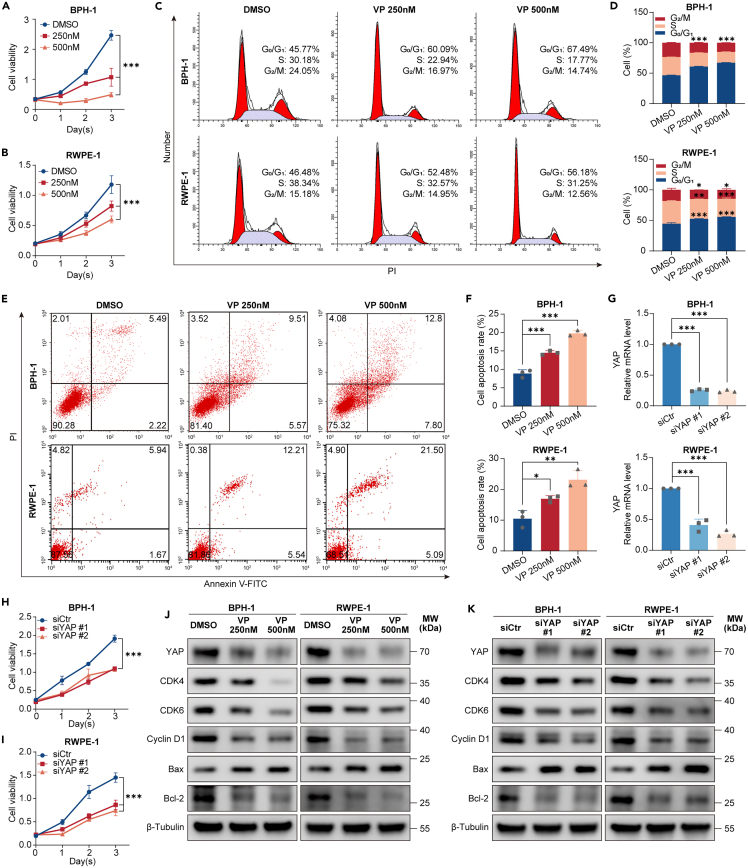
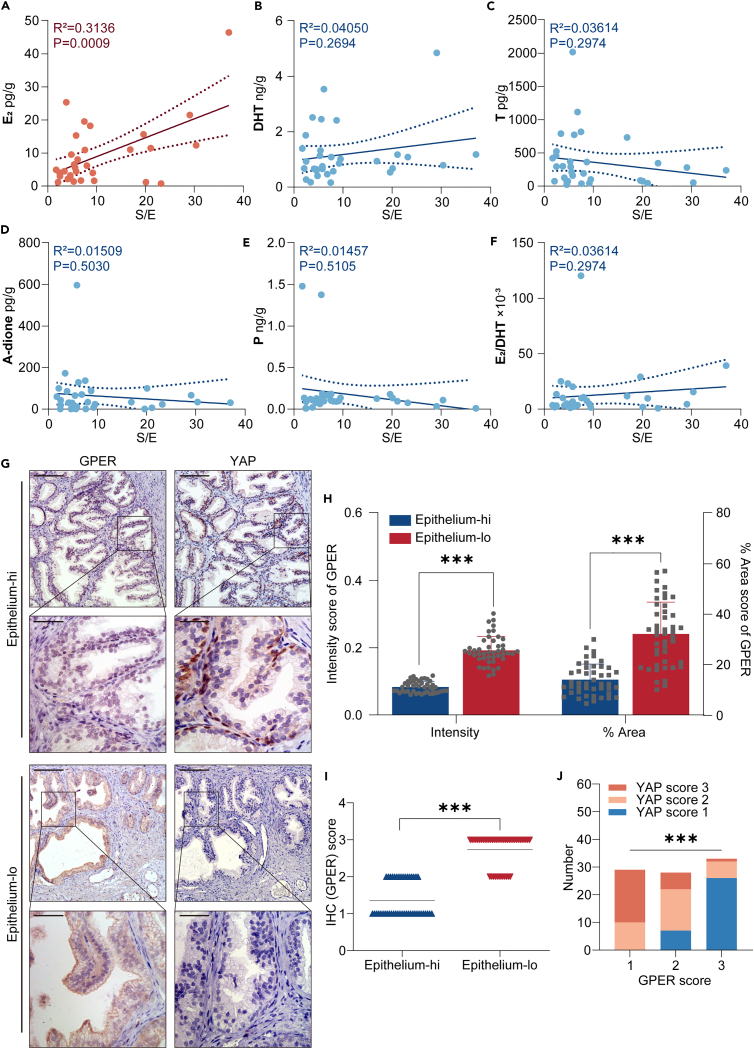
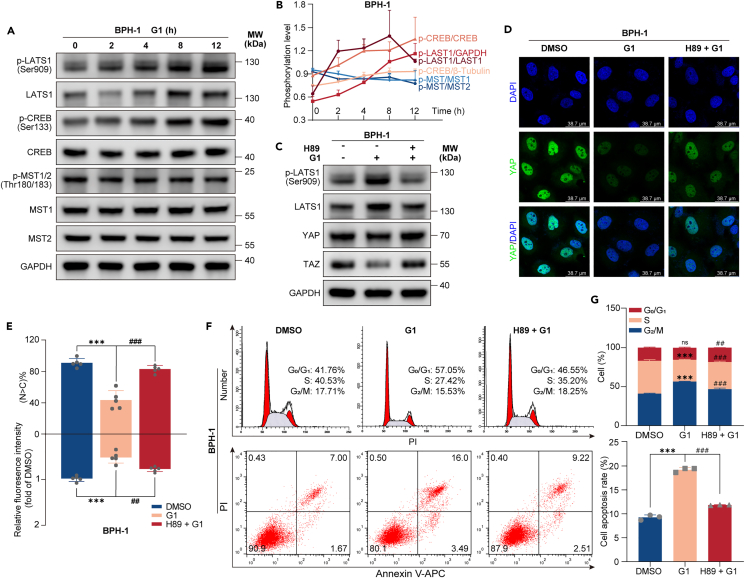
Benign prostatic hyperplasia (BPH) occurs when there is an imbalance between the proliferation and death of prostate cells, which is regulated tightly by estrogen signaling. However, the role of G protein-coupled estrogen receptor (GPER) in prostate cell survival remains ambiguous. In this study, we observed that prostates with epithelial hyperplasia showed increased yes-associated protein 1 (YAP) expression and decreased levels of estrogen and GPER. Blocking YAP through genetic or drug interventions led to reduced proliferation and increased apoptosis in the prostate epithelial cells. Interestingly, GPER agonists produced similar effects. GPER activation enhanced the phosphorylation and degradation of YAP, which was crucial for suppressing cell proliferation and survival. The Gαs/cAMP/PKA/LATS pathway, downstream of GPER, transmitted signals that facilitated YAP inhibition. This study investigated the interaction between GPER and YAP in the prostate epithelial cells and its contribution to BPH development. It lays the groundwork for future research on developing BPH treatments.
Keywords: Biochemistry; Cell biology; Molecular biology; Prostate disease.
© 2024 The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures



Similar articles
-
Identification of the G protein-coupled estrogen receptor (GPER) in human prostate: expression site of the estrogen receptor in the benign and neoplastic gland.Andrology. 2016 Jan;4(1):121-7. doi: 10.1111/andr.12131. Epub 2015 Dec 29. Andrology. 2016. PMID: 26714890
-
The treatment effects of flaxseed-derived secoisolariciresinol diglycoside and its metabolite enterolactone on benign prostatic hyperplasia involve the G protein-coupled estrogen receptor 1.Appl Physiol Nutr Metab. 2016 Dec;41(12):1303-1310. doi: 10.1139/apnm-2016-0332. Epub 2016 Sep 27. Appl Physiol Nutr Metab. 2016. PMID: 27849354
-
G protein coupled estrogen receptor attenuates mechanical stress-mediated apoptosis of chondrocyte in osteoarthritis via suppression of Piezo1.Mol Med. 2021 Aug 28;27(1):96. doi: 10.1186/s10020-021-00360-w. Mol Med. 2021. PMID: 34454425 Free PMC article.
-
17β-Estradiol, through activating the G protein-coupled estrogen receptor, exacerbates the complication of benign prostatic hyperplasia in type 2 diabetes mellitus patients by inducing prostate proliferation.J Pharm Anal. 2024 Sep;14(9):100962. doi: 10.1016/j.jpha.2024.03.003. Epub 2024 Mar 12. J Pharm Anal. 2024. PMID: 39350964 Free PMC article.
-
Expression and Role of the G Protein-Coupled Estrogen Receptor (GPR30/GPER) in the Development and Immune Response in Female Reproductive Cancers.Front Endocrinol (Lausanne). 2020 Aug 20;11:544. doi: 10.3389/fendo.2020.00544. eCollection 2020. Front Endocrinol (Lausanne). 2020. PMID: 32973677 Free PMC article. Review.
Cited by
-
Tracing the Evolution of Sex Hormones and Receptor-Mediated Immune Microenvironmental Differences in Prostate and Bladder Cancers: From Embryonic Development to Disease.Adv Sci (Weinh). 2025 Apr;12(13):e2407715. doi: 10.1002/advs.202407715. Epub 2025 Feb 25. Adv Sci (Weinh). 2025. PMID: 40007149 Free PMC article. Review.
-
Network pharmacology and experimental validation to explore the pharmacological mechanism of saw palmetto and its core ingredients in benign prostatic hyperplasia treatment.Naunyn Schmiedebergs Arch Pharmacol. 2025 Jan;398(1):543-555. doi: 10.1007/s00210-024-03289-z. Epub 2024 Jul 17. Naunyn Schmiedebergs Arch Pharmacol. 2025. PMID: 39017714
-
The miRNAs 203a/210-3p/5001-5p regulate the androgen/androgen receptor/YAP-induced migration in prostate cancer cells.Cancer Med. 2024 Aug;13(16):e70106. doi: 10.1002/cam4.70106. Cancer Med. 2024. PMID: 39149855 Free PMC article.
-
The pathogenesis of benign prostatic hyperplasia and the roles of Prdx3, oxidative stress, pyroptosis and autophagy:a review.Front Oncol. 2025 Aug 5;15:1579539. doi: 10.3389/fonc.2025.1579539. eCollection 2025. Front Oncol. 2025. PMID: 40837016 Free PMC article. Review.
References
LinkOut - more resources
Full Text Sources
Research Materials
