Drug target prediction through deep learning functional representation of gene signatures
- PMID: 38424040
- PMCID: PMC10904399
- DOI: 10.1038/s41467-024-46089-y
Drug target prediction through deep learning functional representation of gene signatures
Abstract
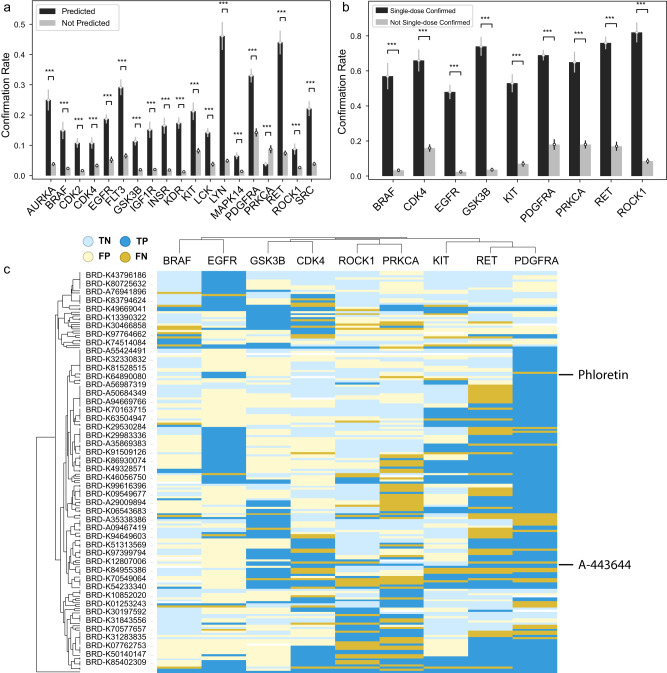
Many machine learning applications in bioinformatics currently rely on matching gene identities when analyzing input gene signatures and fail to take advantage of preexisting knowledge about gene functions. To further enable comparative analysis of OMICS datasets, including target deconvolution and mechanism of action studies, we develop an approach that represents gene signatures projected onto their biological functions, instead of their identities, similar to how the word2vec technique works in natural language processing. We develop the Functional Representation of Gene Signatures (FRoGS) approach by training a deep learning model and demonstrate that its application to the Broad Institute's L1000 datasets results in more effective compound-target predictions than models based on gene identities alone. By integrating additional pharmacological activity data sources, FRoGS significantly increases the number of high-quality compound-target predictions relative to existing approaches, many of which are supported by in silico and/or experimental evidence. These results underscore the general utility of FRoGS in machine learning-based bioinformatics applications. Prediction networks pre-equipped with the knowledge of gene functions may help uncover new relationships among gene signatures acquired by large-scale OMICs studies on compounds, cell types, disease models, and patient cohorts.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures