

The transcription factor ChREBP Orchestrates liver carcinogenesis by coordinating the PI3K/AKT signaling and cancer metabolism
- PMID: 38424041
- PMCID: PMC10904844
- DOI: 10.1038/s41467-024-45548-w
The transcription factor ChREBP Orchestrates liver carcinogenesis by coordinating the PI3K/AKT signaling and cancer metabolism
Abstract
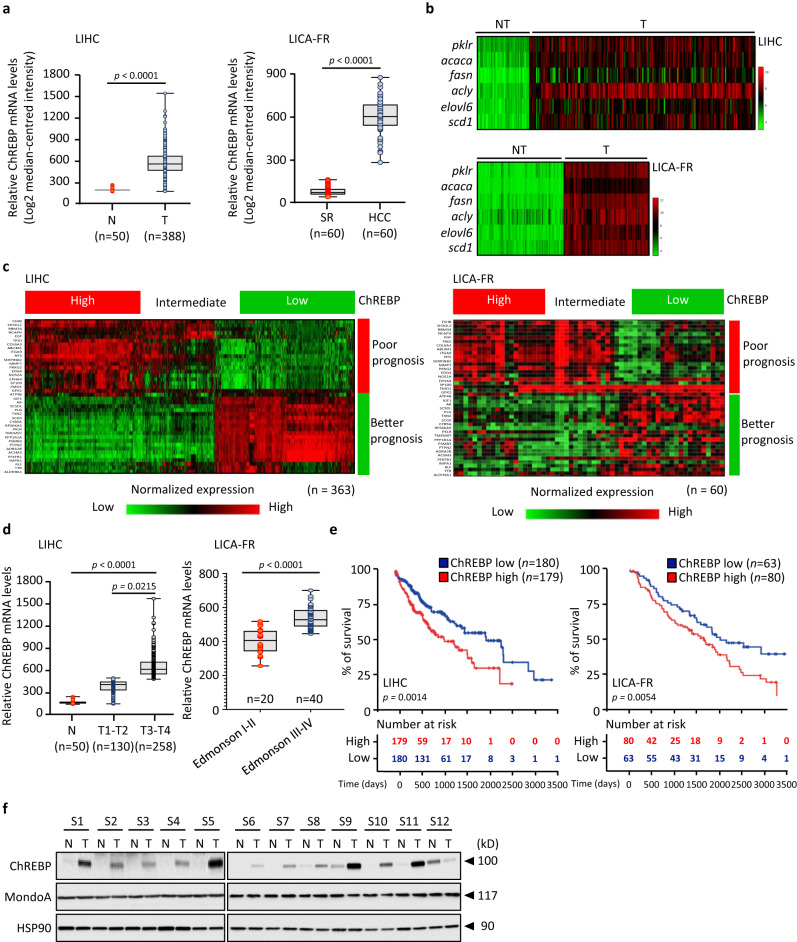
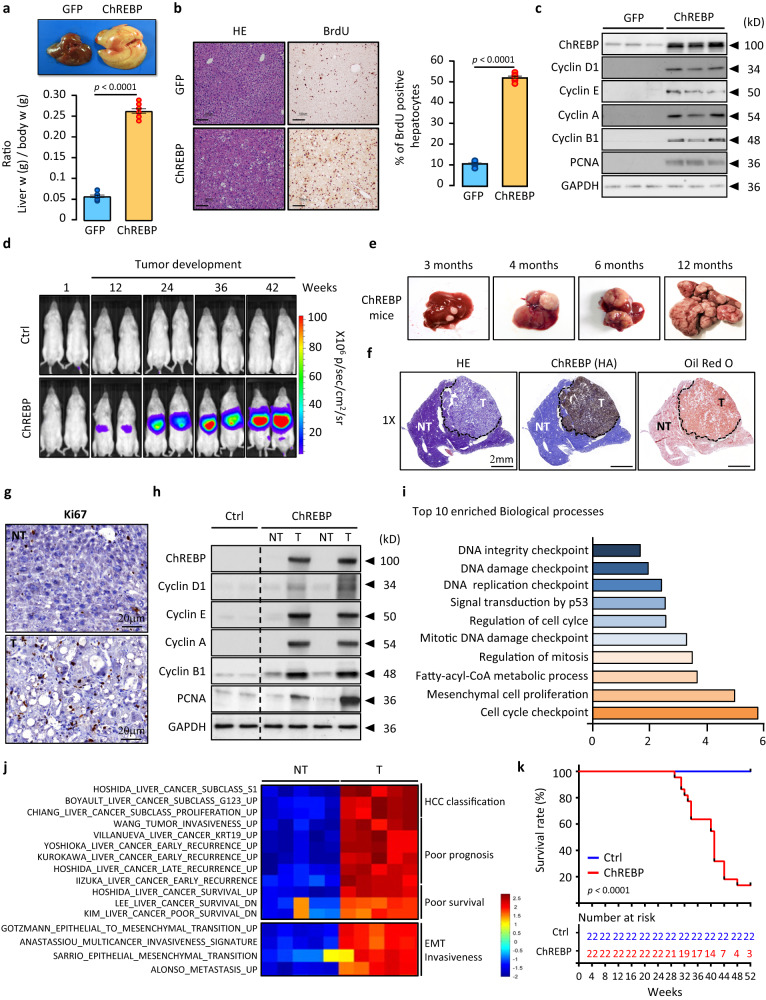
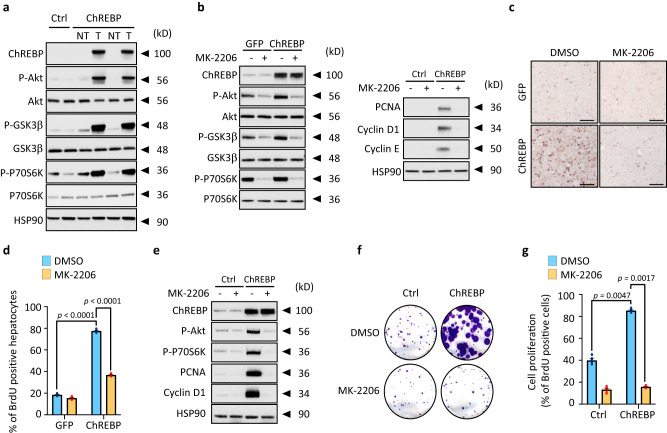
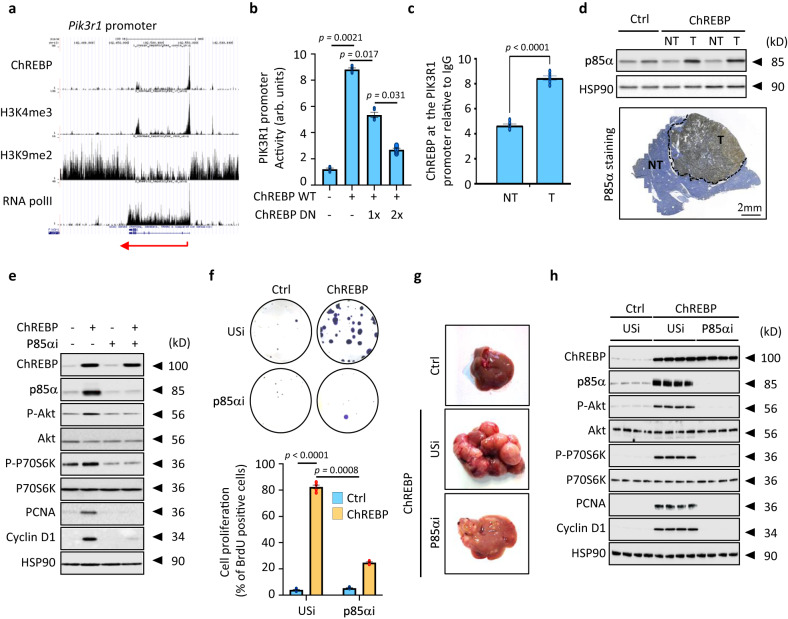
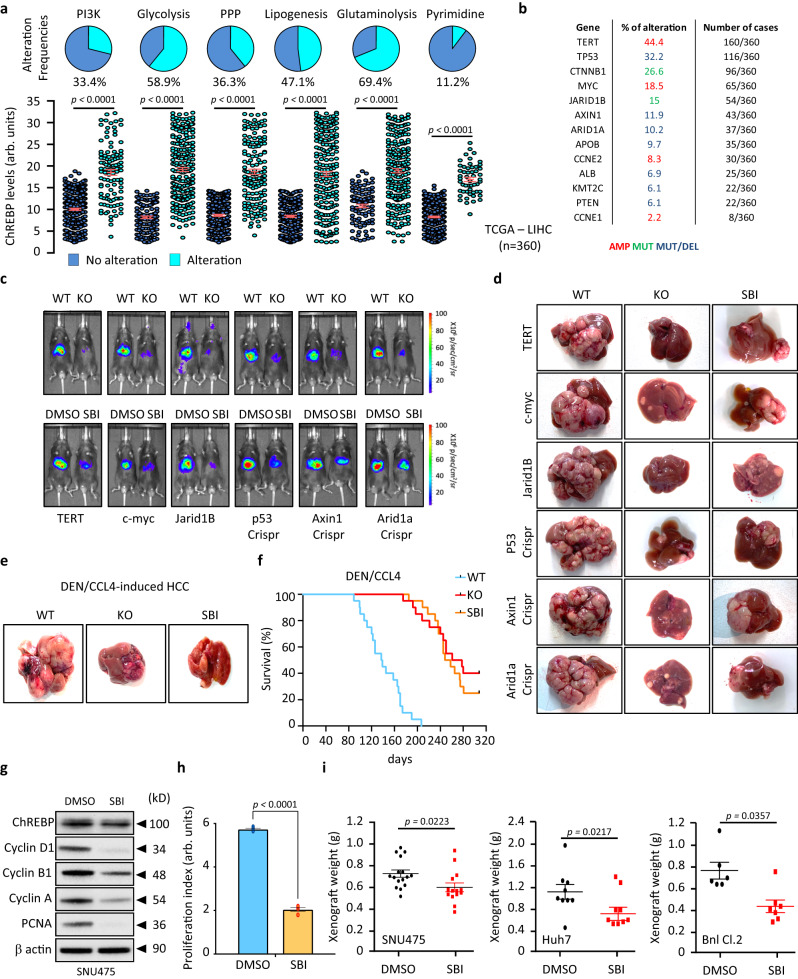
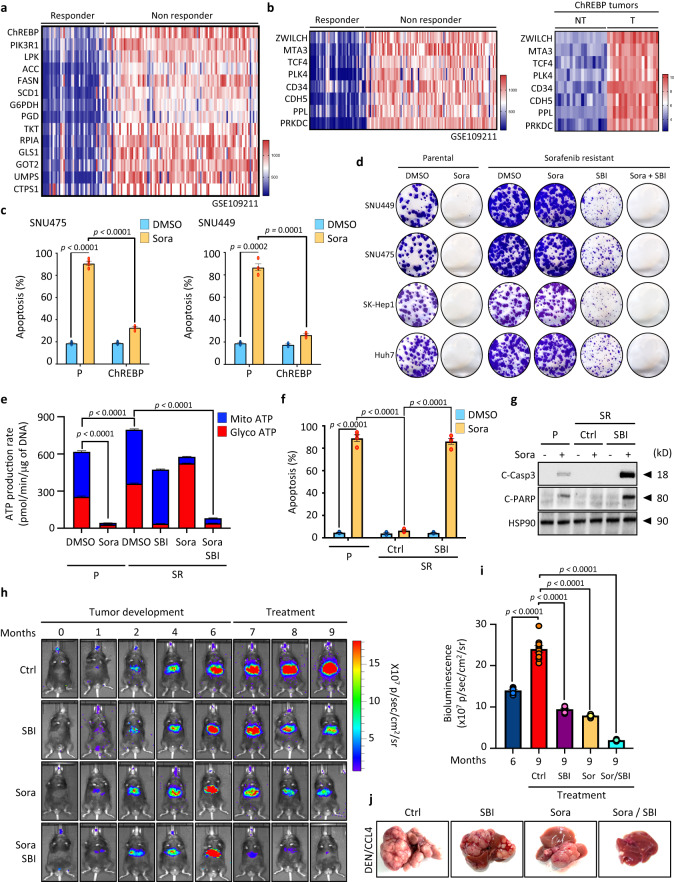
Cancer cells integrate multiple biosynthetic demands to drive unrestricted proliferation. How these cellular processes crosstalk to fuel cancer cell growth is still not fully understood. Here, we uncover the mechanisms by which the transcription factor Carbohydrate responsive element binding protein (ChREBP) functions as an oncogene during hepatocellular carcinoma (HCC) development. Mechanistically, ChREBP triggers the expression of the PI3K regulatory subunit p85α, to sustain the activity of the pro-oncogenic PI3K/AKT signaling pathway in HCC. In parallel, increased ChREBP activity reroutes glucose and glutamine metabolic fluxes into fatty acid and nucleic acid synthesis to support PI3K/AKT-mediated HCC growth. Thus, HCC cells have a ChREBP-driven circuitry that ensures balanced coordination between PI3K/AKT signaling and appropriate cell anabolism to support HCC development. Finally, pharmacological inhibition of ChREBP by SBI-993 significantly suppresses in vivo HCC tumor growth. Overall, we show that targeting ChREBP with specific inhibitors provides an attractive therapeutic window for HCC treatment.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
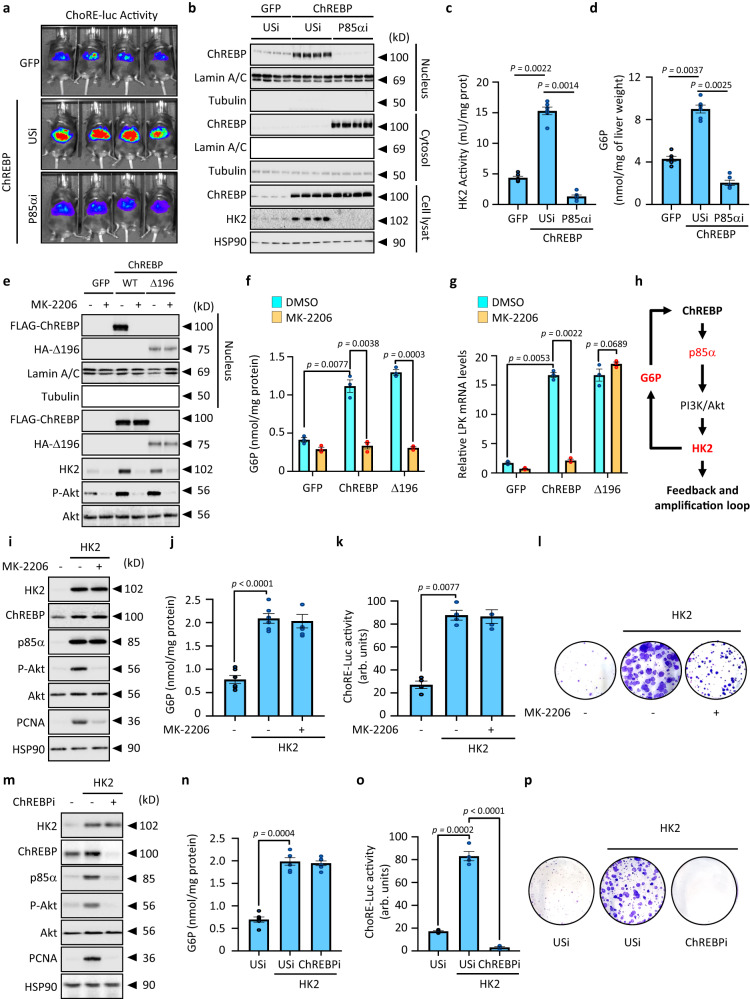
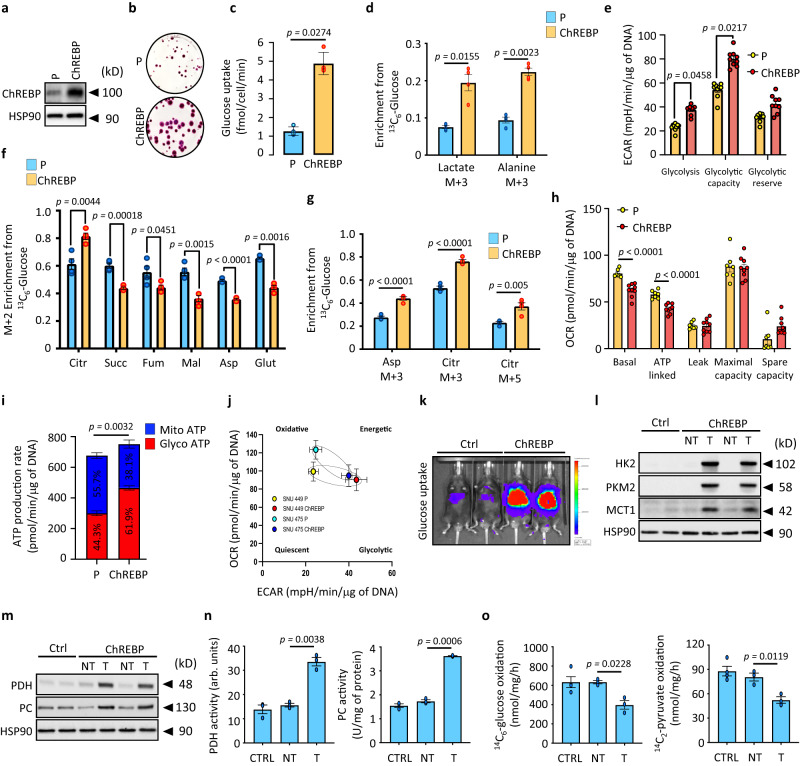
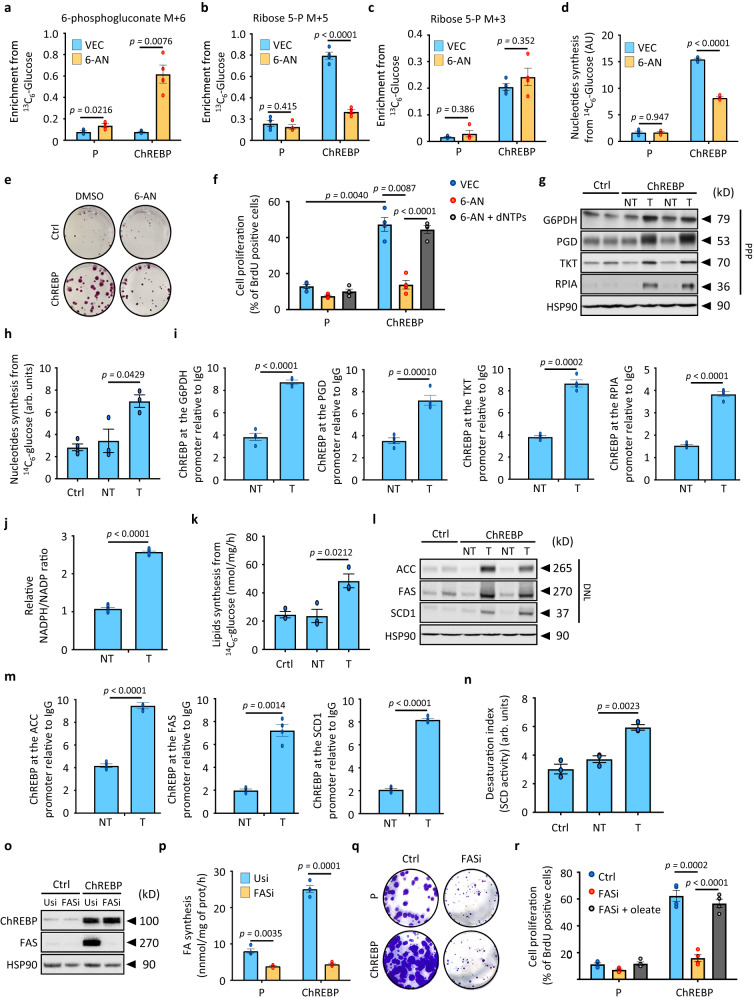
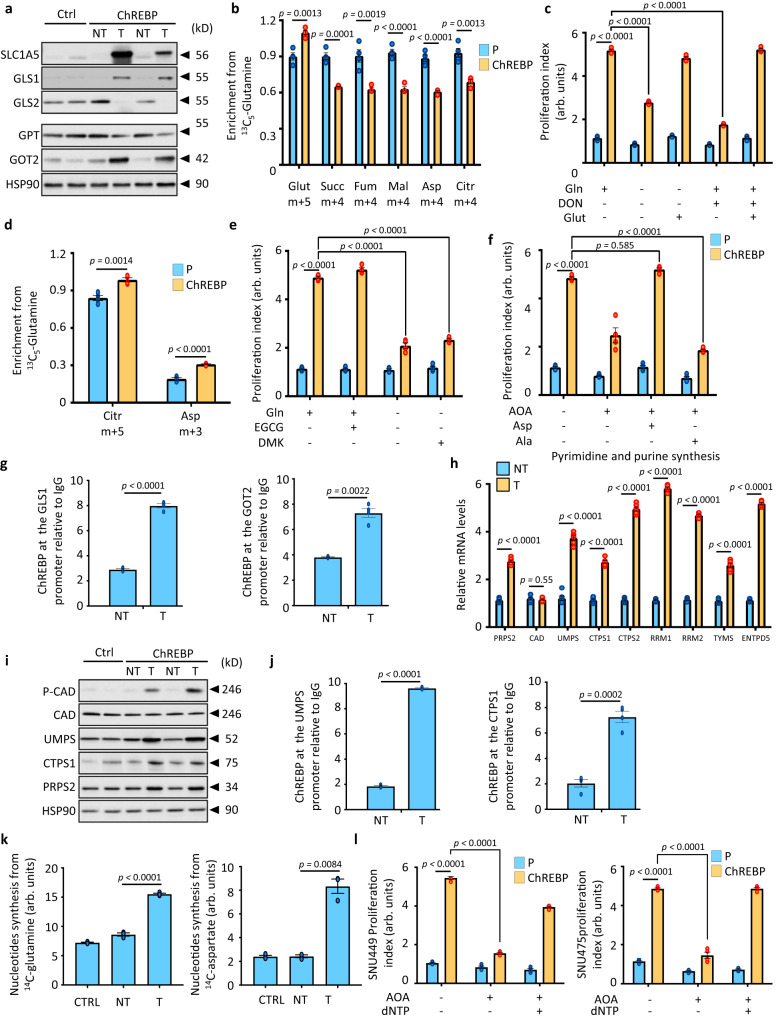
References
MeSH terms
Substances
Grants and funding
- StG-336629/EC | EC Seventh Framework Programm | FP7 Ideas: European Research Council (FP7-IDEAS-ERC - Specific Programme: "Ideas" Implementing the Seventh Framework Programme of the European Community for Research, Technological Development and Demonstration Activities (2007 to 2013))
- RS14/75-89/Ligue Contre le Cancer
- SFI20121205630/Fondation ARC pour la Recherche sur le Cancer (ARC Foundation for Cancer Research)
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
