

Exploring the gut DNA virome in fecal immunochemical test stool samples reveals associations with lifestyle in a large population-based study
- PMID: 38424056
- PMCID: PMC10904388
- DOI: 10.1038/s41467-024-46033-0
Exploring the gut DNA virome in fecal immunochemical test stool samples reveals associations with lifestyle in a large population-based study
Abstract
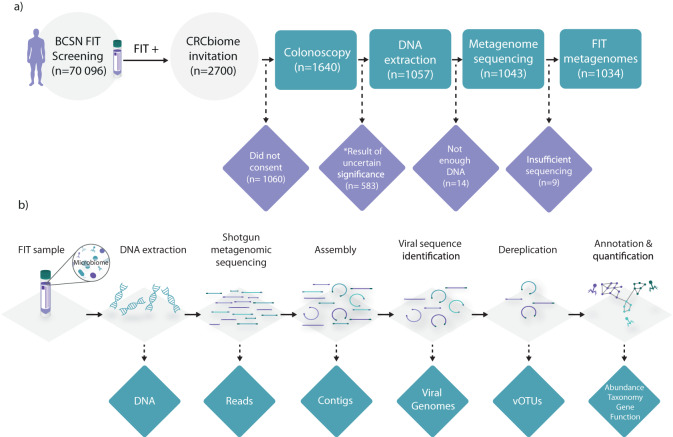
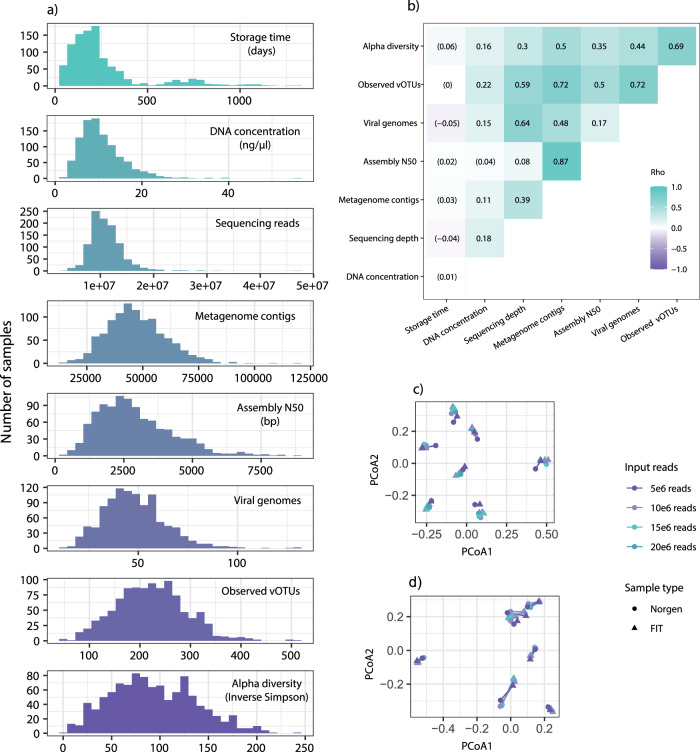
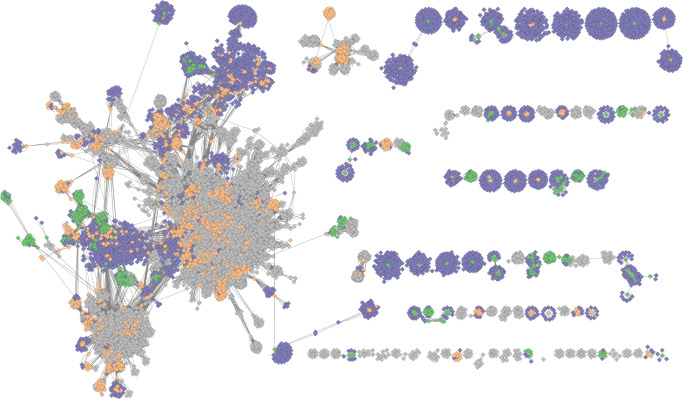
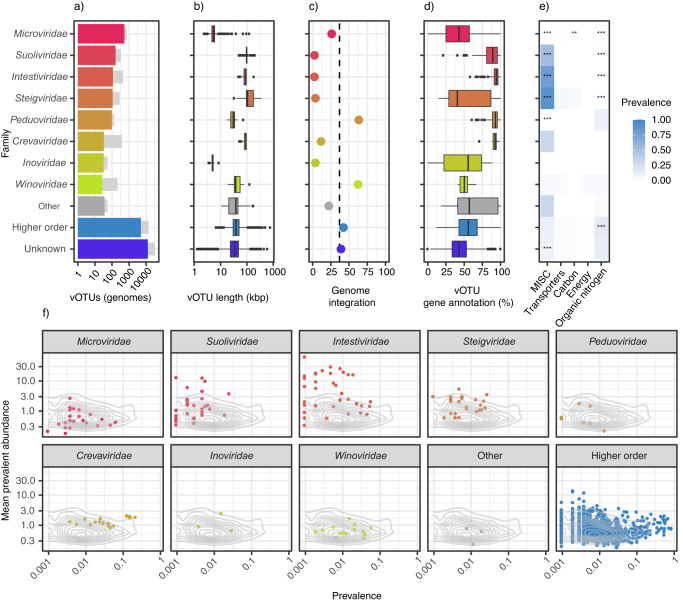
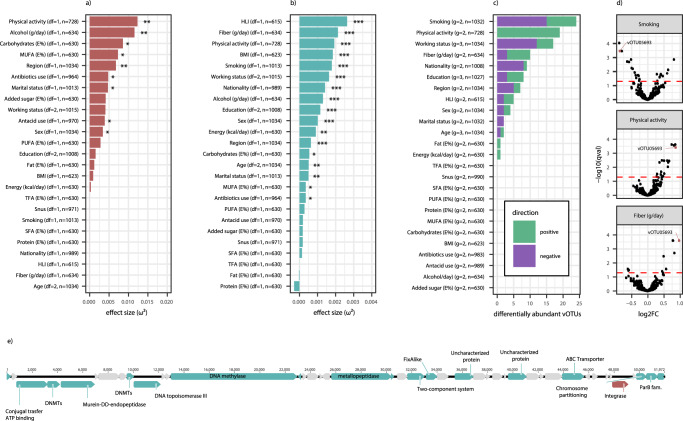
Stool samples for fecal immunochemical tests (FIT) are collected in large numbers worldwide as part of colorectal cancer screening programs. Employing FIT samples from 1034 CRCbiome participants, recruited from a Norwegian colorectal cancer screening study, we identify, annotate and characterize more than 18000 DNA viruses, using shotgun metagenome sequencing. Only six percent of them are assigned to a known taxonomic family, with Microviridae being the most prevalent viral family. Linking individual profiles to comprehensive lifestyle and demographic data shows 17/25 of the variables to be associated with the gut virome. Physical activity, smoking, and dietary fiber consumption exhibit strong and consistent associations with both diversity and relative abundance of individual viruses, as well as with enrichment for auxiliary metabolic genes. We demonstrate the suitability of FIT samples for virome analysis, opening an opportunity for large-scale studies of this enigmatic part of the gut microbiome. The diverse viral populations and their connections to the individual lifestyle uncovered herein paves the way for further exploration of the role of the gut virome in health and disease.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
