Brain function in classic galactosemia, a galactosemia network (GalNet) members review
- PMID: 38425716
- PMCID: PMC10902464
- DOI: 10.3389/fgene.2024.1355962
Brain function in classic galactosemia, a galactosemia network (GalNet) members review
Abstract
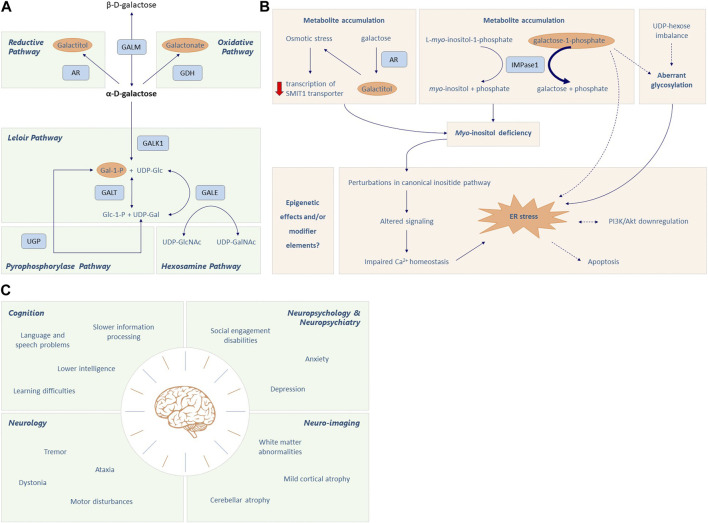
Classic galactosemia (CG, OMIM #230400, ORPHA: 79,239) is a hereditary disorder of galactose metabolism that, despite treatment with galactose restriction, affects brain function in 85% of the patients. Problems with cognitive function, neuropsychological/social emotional difficulties, neurological symptoms, and abnormalities in neuroimaging and electrophysiological assessments are frequently reported in this group of patients, with an enormous individual variability. In this review, we describe the role of impaired galactose metabolism on brain dysfunction based on state of the art knowledge. Several proposed disease mechanisms are discussed, as well as the time of damage and potential treatment options. Furthermore, we combine data from longitudinal, cross-sectional and retrospective studies with the observations of specialist teams treating this disease to depict the brain disease course over time. Based on current data and insights, the majority of patients do not exhibit cognitive decline. A subset of patients, often with early onset cerebral and cerebellar volume loss, can nevertheless experience neurological worsening. While a large number of patients with CG suffer from anxiety and depression, the increased complaints about memory loss, anxiety and depression at an older age are likely multifactorial in origin.
Keywords: brain; classic galactosemia; cognitive problems; galactose; movement disorders; neurodevelopment; neuropsychiatry.
Copyright © 2024 Panis, Vos, Barić, Bosch, Brouwers, Burlina, Cassiman, Coman, Couce, Das, Demirbas, Empain, Gautschi, Grafakou, Grunewald, Kingma, Knerr, Leão-Teles, Möslinger, Murphy, Õunap, Pané, Paci, Parini, Rivera, Scholl-Bürgi, Schwartz, Sdogou, Shakerdi, Skouma, Stepien, Treacy, Waisbren, Berry and Rubio-Gozalbo.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Figures