

Case report: Dihydropyridine receptor (CACNA1S) congenital myopathy, a novel phenotype with early onset periodic paralysis
- PMID: 38426167
- PMCID: PMC10902085
- DOI: 10.3389/fneur.2024.1359479
Case report: Dihydropyridine receptor (CACNA1S) congenital myopathy, a novel phenotype with early onset periodic paralysis
Abstract
Introduction: CACNA1S related congenital myopathy is an emerging recently described entity. In this report we describe 2 sisters with mutations in the CACNA1S gene and the novel phenotype of congenital myopathy and infantile onset episodic weakness.
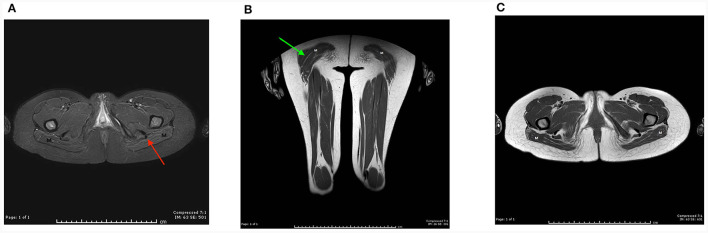
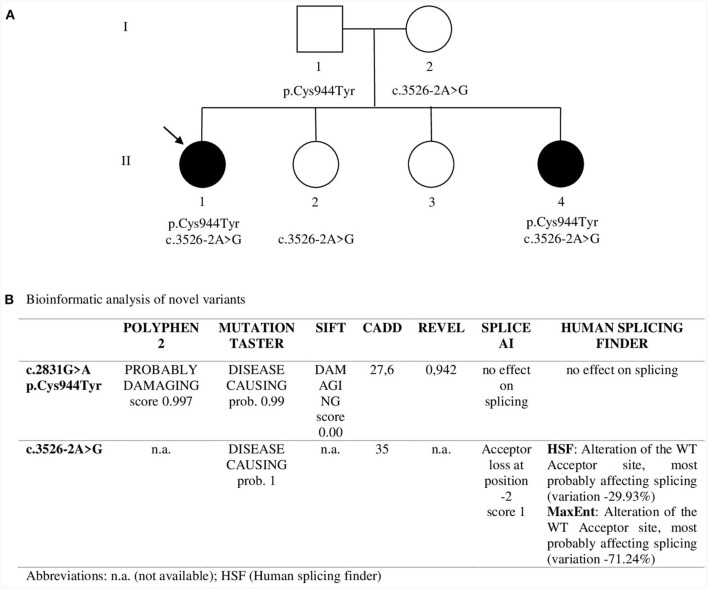
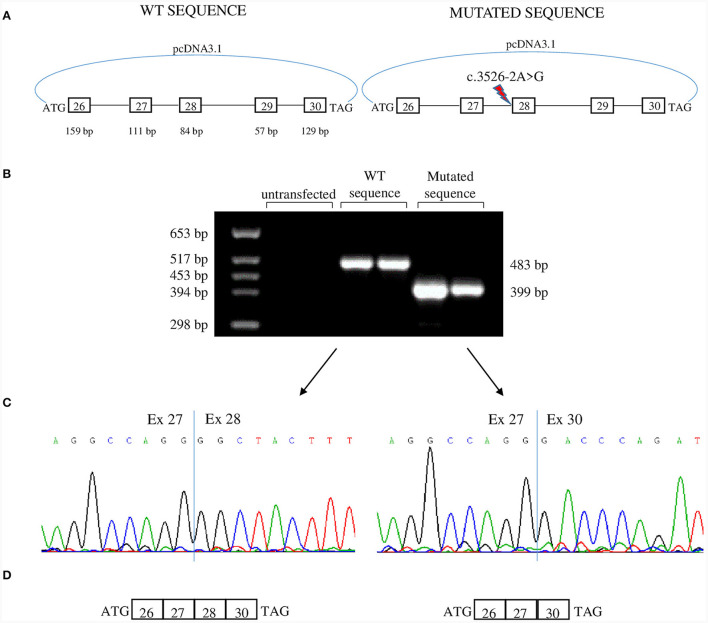
Clinical description: Both sisters had neonatal onset hypotonia, muscle weakness, and delayed walking. Episodic weakness started in infancy and continued thereafter, provoked mostly by cold exposure. Muscle imaging revealed fat replacement of gluteus maximus muscles. Next generation sequencing found the missense p.Cys944Tyr variant and the novel splicing variant c.3526-2A>G in CACNA1S. Minigene assay revealed the splicing variant caused skipping of exon 28 from the transcript, potentially affecting protein folding and/or voltage dependent activation.
Conclusion: This novel phenotype supports the notion that there are age related differences in the clinical expression of CACNA1S gene mutations. This expands our understanding of mutations located in regions of the CACNA1S outside the highly conserved S4 segment, where most mutations thus far have been identified.
Keywords: CACNA1S; Cav1.1; DHPR; congenital myopathy; episodic weakness; novel phenotype; periodic paralysis; splice minigene assay.
Copyright © 2024 Aburahma, Rousan, Shboul, Biella, Lucchiari, Comi, Meola and Pagliarani.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Figures
Similar articles
-
Dihydropyridine receptor (DHPR, CACNA1S) congenital myopathy.Acta Neuropathol. 2017 Apr;133(4):517-533. doi: 10.1007/s00401-016-1656-8. Epub 2016 Dec 23. Acta Neuropathol. 2017. PMID: 28012042
-
CACNA1S-associated triadopathy presenting with myalgia, muscle weakness, and asymptomatic hyperCKemia.Ther Adv Neurol Disord. 2025 Feb 26;18:17562864251317961. doi: 10.1177/17562864251317961. eCollection 2025. Ther Adv Neurol Disord. 2025. PMID: 40018084 Free PMC article.
-
A novel CACNA1S gene variant in a child with hypokalemic periodic paralysis: a case report and literature review.BMC Pediatr. 2023 Oct 2;23(1):500. doi: 10.1186/s12887-023-04326-1. BMC Pediatr. 2023. PMID: 37784084 Free PMC article. Review.
-
Core myopathy in two siblings with a biallelic variant in the CACNA1S gene-A case series study.Clin Case Rep. 2024 Aug 5;12(8):e9251. doi: 10.1002/ccr3.9251. eCollection 2024 Aug. Clin Case Rep. 2024. PMID: 39104734 Free PMC article.
-
A novel de novo COL6A1 mutation emphasizes the role of intron 14 donor splice site defects as a cause of moderate-progressive form of ColVI myopathy - a case report and review of the genotype-phenotype correlation.Folia Neuropathol. 2017;55(3):214-220. doi: 10.5114/fn.2017.70486. Folia Neuropathol. 2017. PMID: 28984114 Review.