

Genetics of Gallstone Disease and Their Clinical Significance: A Narrative Review
- PMID: 38426197
- PMCID: PMC10899874
- DOI: 10.14218/JCTH.2023.00563
Genetics of Gallstone Disease and Their Clinical Significance: A Narrative Review
Abstract
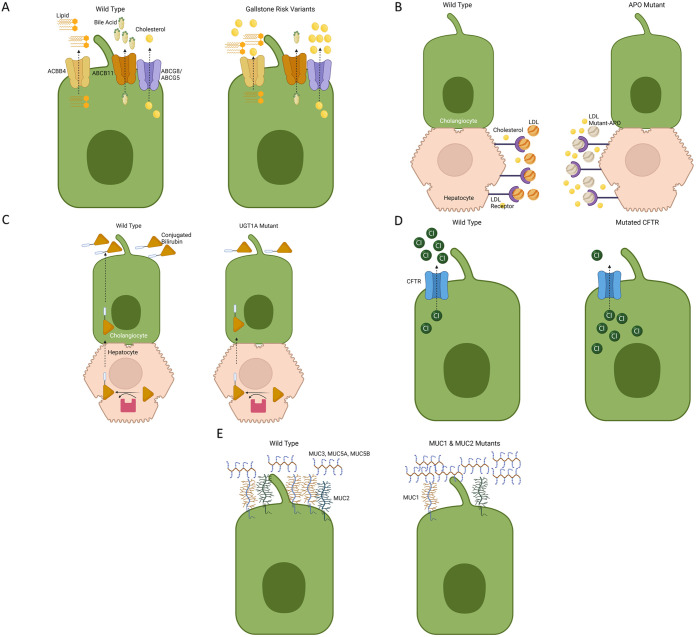
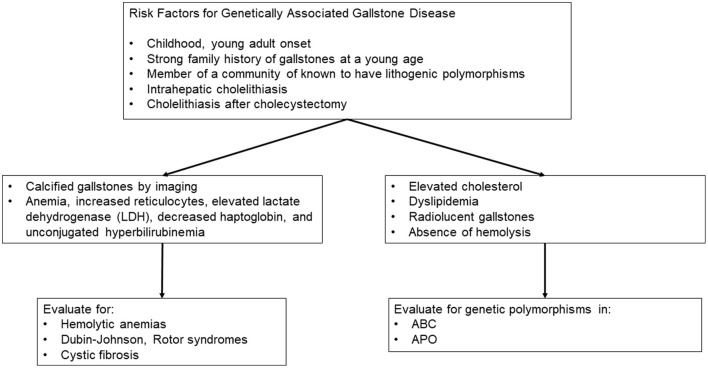
Gallstone (GS) disease is common and arises from a combination of genetic and environmental factors. Although genetic abnormalities specifically leading to cholesterol GSs are rare, there are clinically significant gene variants associated with cholesterol GSs. In contrast, most bilirubin GSs can be attributed to genetic defects. The pathogenesis of cholesterol and bilirubin GSs differs greatly. Cholesterol GSs are notably influenced by genetic variants within the ABC protein superfamily, including ABCG8, ABCG5, ABCB4, and ABCB11, as well as genes from the apolipoprotein family such as ApoB100 and ApoE (especially the E3/E3 and E3/E4 variants), and members of the MUC family. Conversely, bilirubin GSs are associated with genetic variants in highly expressed hepatic genes, notably UGT1A1, ABCC2 (MRP2), ABCC3 (MRP3), CFTR, and MUC, alongside genetic defects linked to hemolytic anemias and conditions impacting erythropoiesis. While genetic cases constitute a small portion of GS disease, recognizing genetic predisposition is essential for proper diagnosis, treatment, and genetic counseling.
Keywords: ABCG8 protein; ATP-binding cassette transporters; Cholelithiasis; Gallstones; Human; UDP-glucuronosyltransferase A1.
© 2024 Authors.
Conflict of interest statement
GYW has been an Editor-in-Chief of Journal of Clinical and Translational Hepatology since 2013. The other authors have no conflict of interests related to this publication.
Figures
Similar articles
-
Loci from a genome-wide analysis of bilirubin levels are associated with gallstone risk and composition.Gastroenterology. 2010 Dec;139(6):1942-1951.e2. doi: 10.1053/j.gastro.2010.09.003. Epub 2010 Sep 15. Gastroenterology. 2010. PMID: 20837016
-
Increased gallstone risk in humans conferred by common variant of hepatic ATP-binding cassette transporter for cholesterol.Hepatology. 2007 Sep;46(3):793-801. doi: 10.1002/hep.21847. Hepatology. 2007. PMID: 17626266
-
Mice overexpressing hepatic Abcb11 rapidly develop cholesterol gallstones.Mamm Genome. 2005 Dec;16(12):903-8. doi: 10.1007/s00335-004-2465-2. Epub 2005 Dec 8. Mamm Genome. 2005. PMID: 16341669
-
Polymorphisms in ABCG5/G8 transporters linked to hypercholesterolemia and gallstone disease.Nutr Rev. 2008 Jun;66(6):343-8. doi: 10.1111/j.1753-4887.2008.00042.x. Nutr Rev. 2008. PMID: 18522623 Review.
-
Pathogenesis of cholesterol and pigment gallstones: an update.Clin Res Hepatol Gastroenterol. 2011 Apr;35(4):281-7. doi: 10.1016/j.clinre.2011.01.009. Epub 2011 Feb 25. Clin Res Hepatol Gastroenterol. 2011. PMID: 21353662 Review.
Cited by
-
A family with gallstone disease: defining inherited risk in the era of clinical genetic testing.Intern Emerg Med. 2025 Mar;20(2):509-514. doi: 10.1007/s11739-024-03854-7. Epub 2025 Jan 9. Intern Emerg Med. 2025. PMID: 39786488
-
Cardiometabolic index as a predictor of gallstone incidence in U.S. adults: insights from NHANES 2017-2020.BMC Gastroenterol. 2025 Jan 29;25(1):45. doi: 10.1186/s12876-025-03642-3. BMC Gastroenterol. 2025. PMID: 39881275 Free PMC article.
-
Gallstones in the Era of Metabolic Syndrome: Pathophysiology, Risk Prediction, and Management.Cureus. 2025 Mar 13;17(3):e80541. doi: 10.7759/cureus.80541. eCollection 2025 Mar. Cureus. 2025. PMID: 40225487 Free PMC article. Review.
-
Genetics of Gallstones.Genes (Basel). 2025 Feb 22;16(3):256. doi: 10.3390/genes16030256. Genes (Basel). 2025. PMID: 40149408 Free PMC article. Review.
-
C-reactive Protein and Biliary Complications as Independent Predictors of Hospital Stay in Acute Cholecystitis.Cureus. 2025 Jul 9;17(7):e87598. doi: 10.7759/cureus.87598. eCollection 2025 Jul. Cureus. 2025. PMID: 40786322 Free PMC article.
References
Publication types
LinkOut - more resources
Full Text Sources
Miscellaneous