

Adjustment of scRNA-seq data to improve cell-type decomposition of spatial transcriptomics
- PMID: 38426323
- PMCID: PMC10939420
- DOI: 10.1093/bib/bbae063
Adjustment of scRNA-seq data to improve cell-type decomposition of spatial transcriptomics
Erratum in
-
Correction to: Adjustment of scRNA-seq data to improve cell-type decomposition of spatial transcriptomics.Brief Bioinform. 2024 Mar 27;25(3):bbae155. doi: 10.1093/bib/bbae155. Brief Bioinform. 2024. PMID: 38581651 Free PMC article. No abstract available.
Abstract
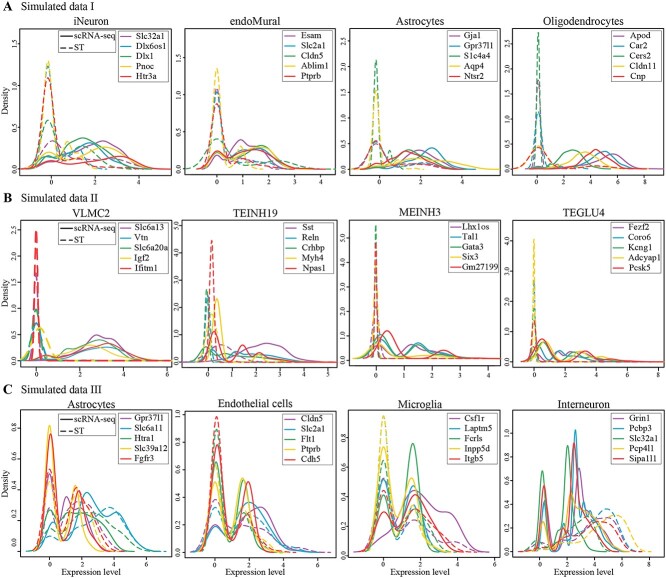
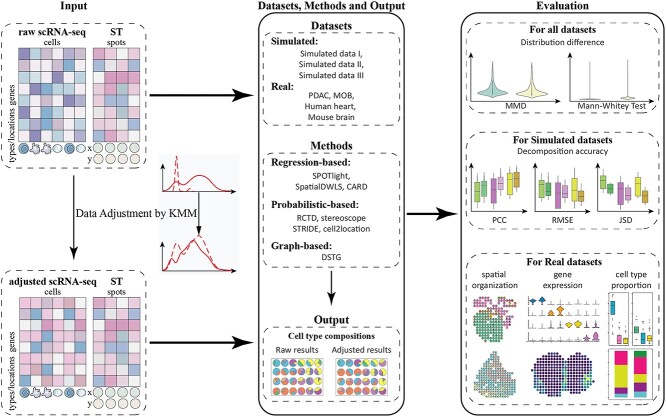
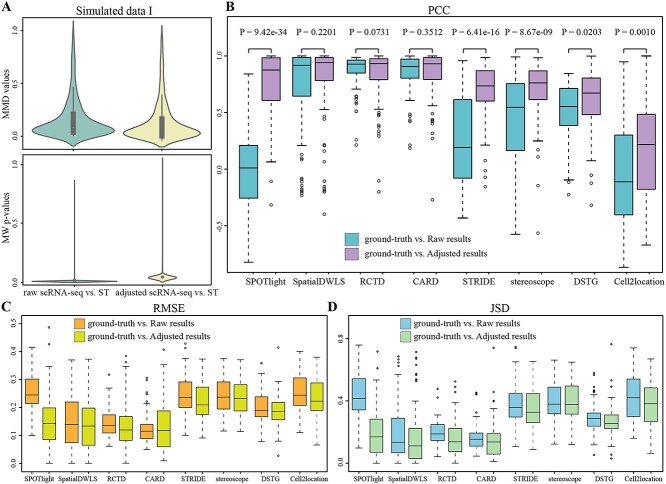
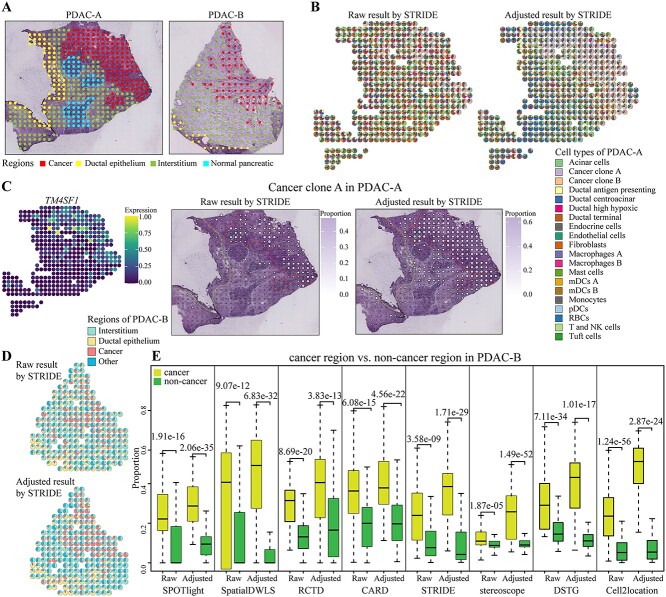
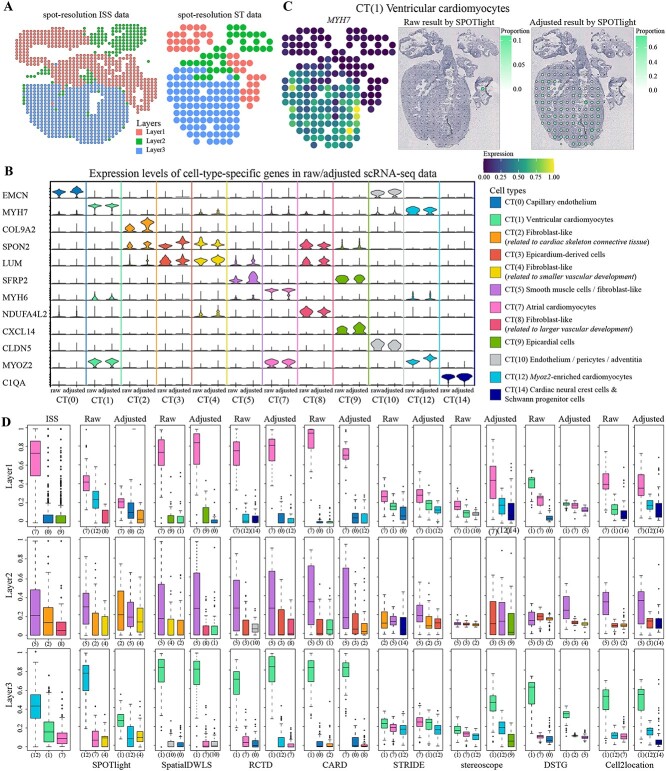
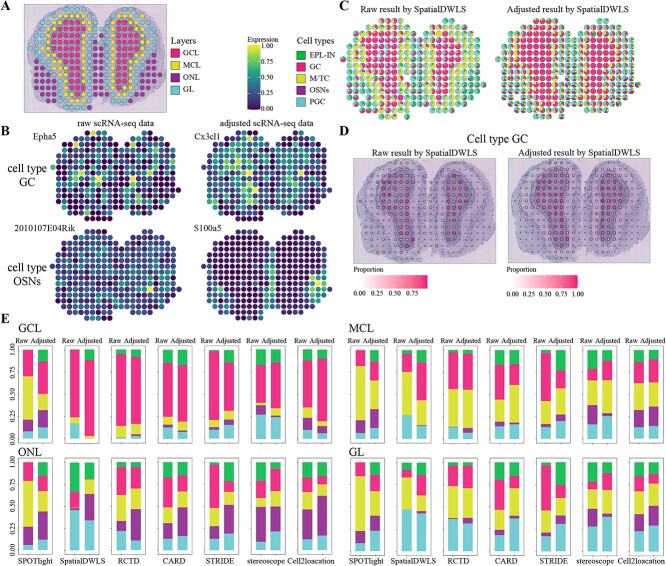
Most sequencing-based spatial transcriptomics (ST) technologies do not achieve single-cell resolution where each captured location (spot) may contain a mixture of cells from heterogeneous cell types, and several cell-type decomposition methods have been proposed to estimate cell type proportions of each spot by integrating with single-cell RNA sequencing (scRNA-seq) data. However, these existing methods did not fully consider the effect of distribution difference between scRNA-seq and ST data for decomposition, leading to biased cell-type-specific genes derived from scRNA-seq for ST data. To address this issue, we develop an instance-based transfer learning framework to adjust scRNA-seq data by ST data to correctly match cell-type-specific gene expression. We evaluate the effect of raw and adjusted scRNA-seq data on cell-type decomposition by eight leading decomposition methods using both simulated and real datasets. Experimental results show that data adjustment can effectively reduce distribution difference and improve decomposition, thus enabling for a more precise depiction on spatial organization of cell types. We highlight the importance of data adjustment in integrative analysis of scRNA-seq with ST data and provide guidance for improved cell-type decomposition.
Keywords: cell-type decomposition; cell-type-specific gene; data adjustment; spatial transcriptomics.
© The Author(s) 2024. Published by Oxford University Press.
Figures
References
-
- Marx V. Method of the Year 2020: spatially resolved transcriptomics. Nat Methods 2021;18:9–14. - PubMed
