

Epistatic pathways can drive HIV-1 escape from integrase strand transfer inhibitors
- PMID: 38427738
- PMCID: PMC10906922
- DOI: 10.1126/sciadv.adn0042
Epistatic pathways can drive HIV-1 escape from integrase strand transfer inhibitors
Abstract
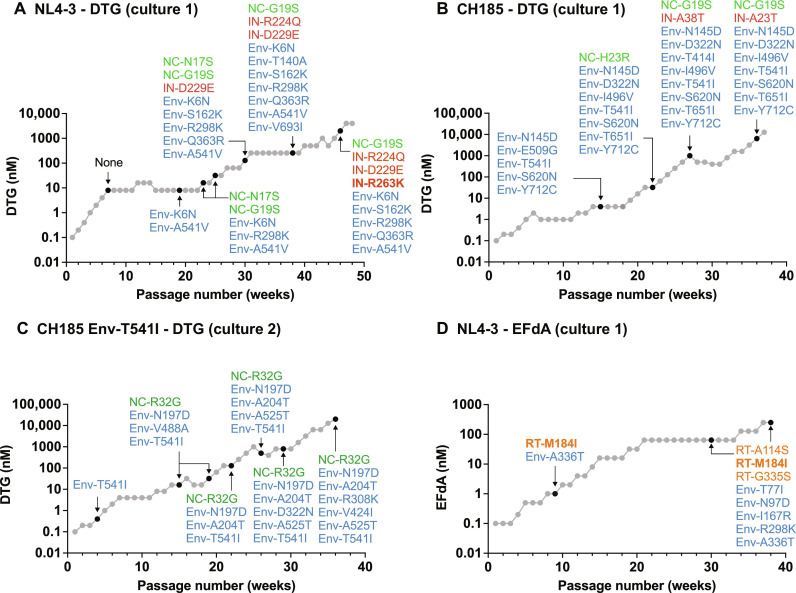
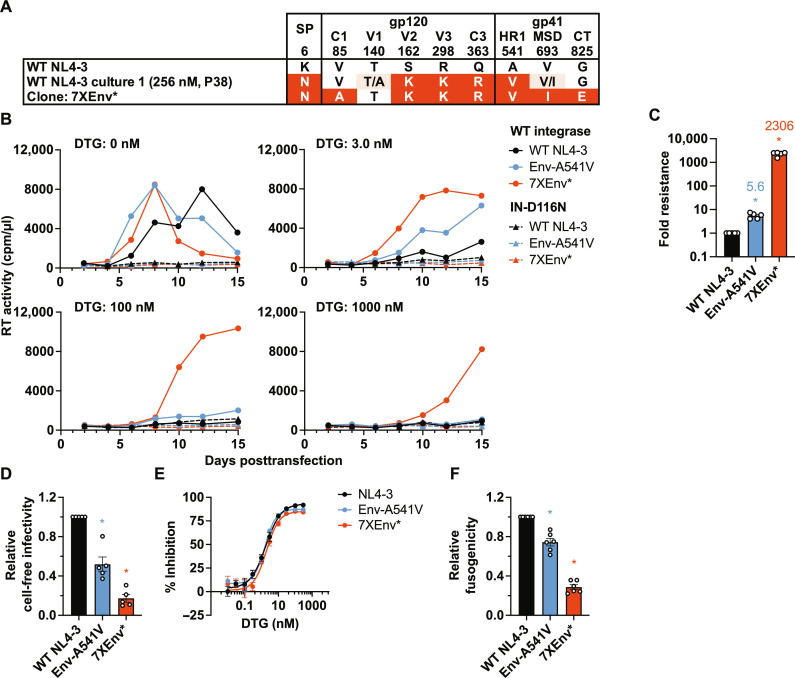
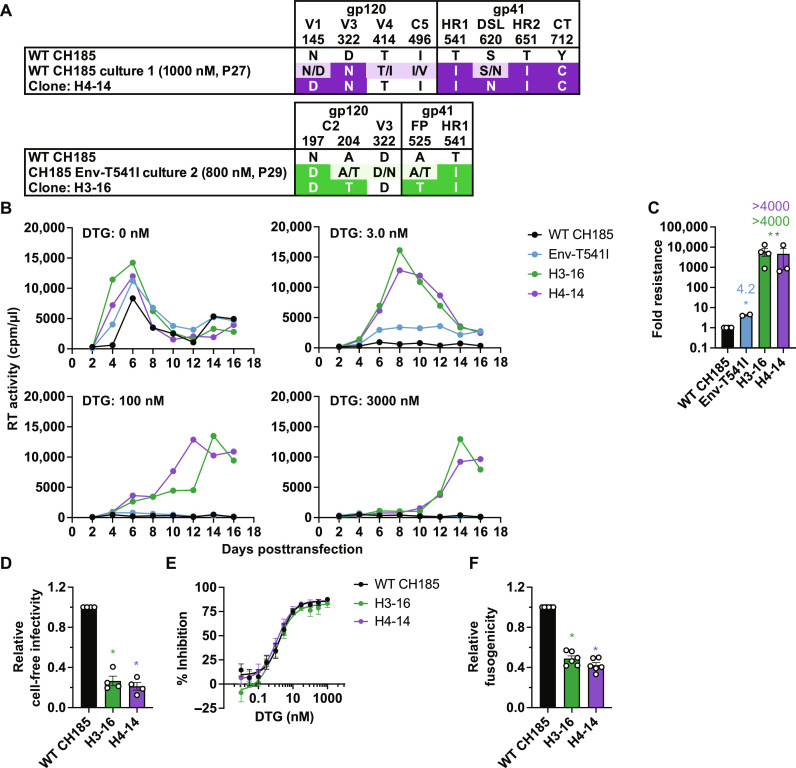
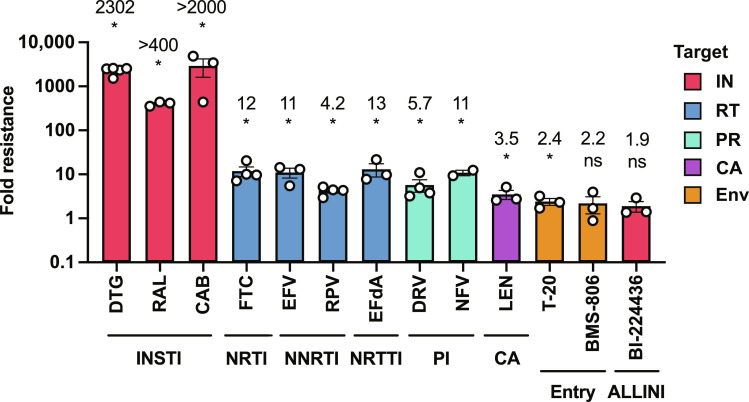
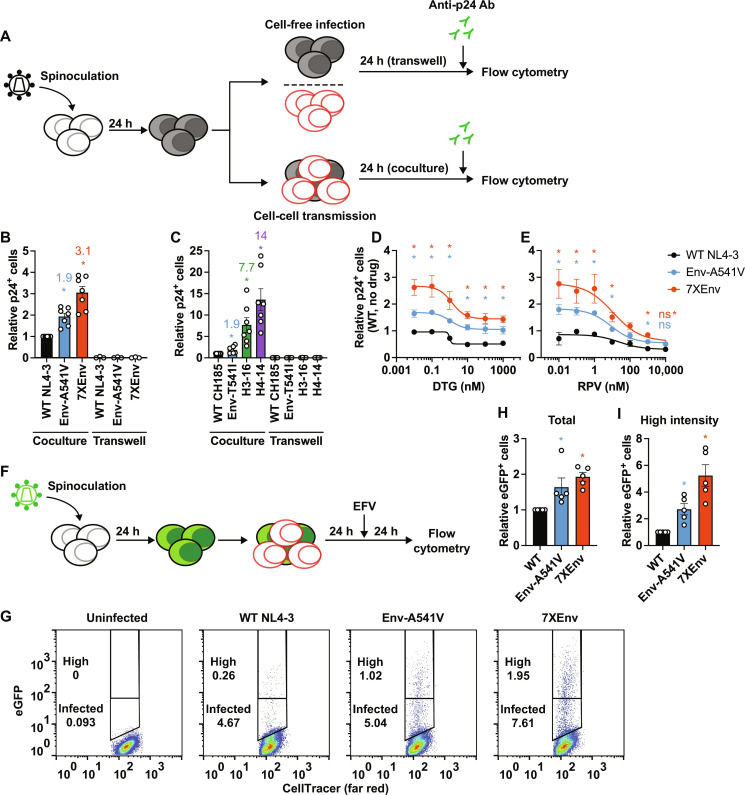
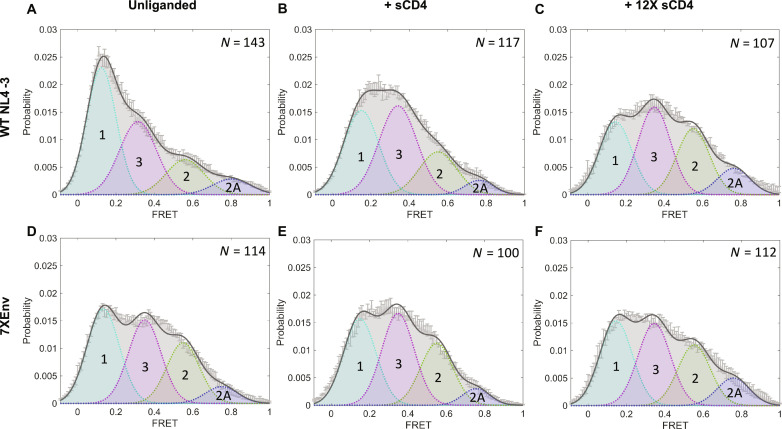
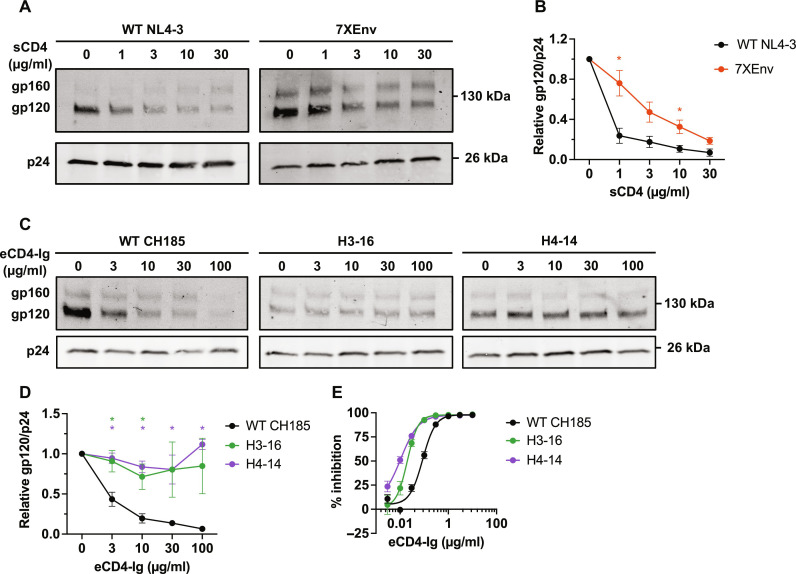
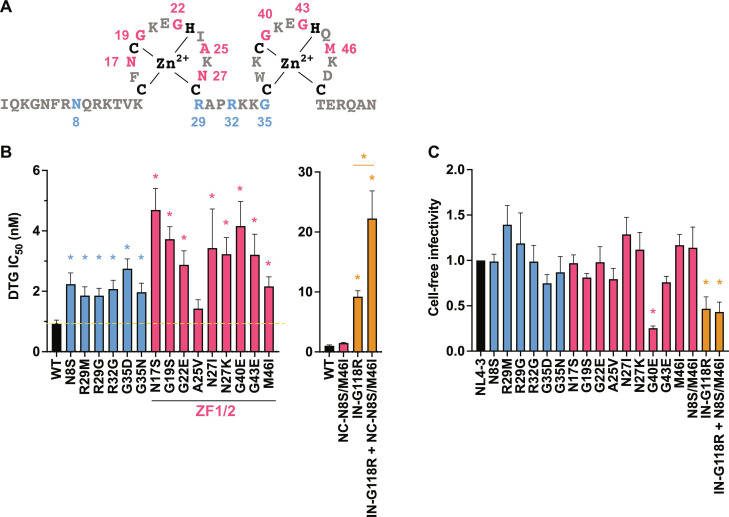
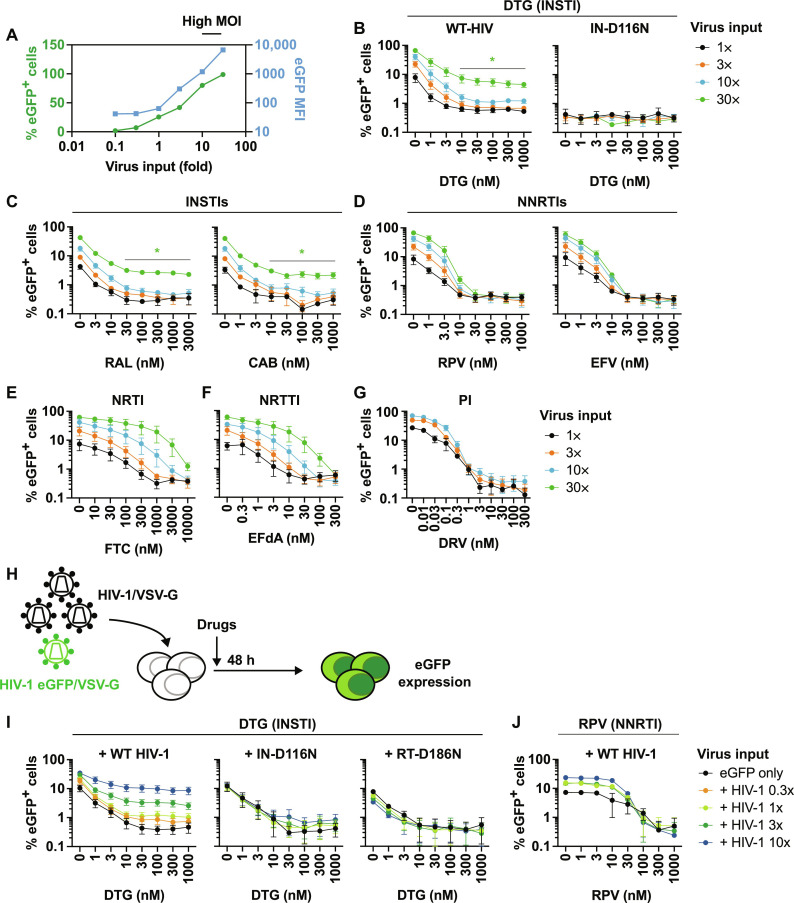
People living with human immunodeficiency virus (HIV) receiving integrase strand transfer inhibitors (INSTIs) have been reported to experience virological failure in the absence of resistance mutations in integrase. To elucidate INSTI resistance mechanisms, we propagated HIV-1 in the presence of escalating concentrations of the INSTI dolutegravir. HIV-1 became resistant to dolutegravir by sequentially acquiring mutations in the envelope glycoprotein (Env) and the nucleocapsid protein. The selected Env mutations enhance the ability of the virus to spread via cell-cell transfer, thereby increasing the multiplicity of infection (MOI). While the selected Env mutations confer broad resistance to multiple classes of antiretrovirals, the fold resistance is ~2 logs higher for INSTIs than for other classes of drugs. We demonstrate that INSTIs are more readily overwhelmed by high MOI than other classes of antiretrovirals. Our findings advance the understanding of how HIV-1 can evolve resistance to antiretrovirals, including the potent INSTIs, in the absence of drug-target gene mutations.
Figures

References
-
- T. R. Kemnic, P. G. Gulick, HIV Antiretroviral Therapy (StatPearls Publishing, 2022). - PubMed
-
- Link J. O., Rhee M. S., Tse W. C., Zheng J., Somoza J. R., Rowe W., Begley R., Chiu A., Mulato A., Hansen D., Singer E., Tsai L. K., Bam R. A., Chou C. H., Canales E., Brizgy G., Zhang J. R., Li J., Graupe M., Morganelli P., Liu Q., Wu Q., Halcomb R. L., Saito R. D., Schroeder S. D., Lazerwith S. E., Bondy S., Jin D., Hung M., Novikov N., Liu X., Villasenor A. G., Cannizzaro C. E., Hu E. Y., Anderson R. L., Appleby T. C., Lu B., Mwangi J., Liclican A., Niedziela-Majka A., Papalia G. A., Wong M. H., Leavitt S. A., Xu Y., Koditek D., Stepan G. J., Yu H., Pagratis N., Clancy S., Ahmadyar S., Cai T. Z., Sellers S., Wolckenhauer S. A., Ling J., Callebaut C., Margot N., Ram R. R., Liu Y. P., Hyland R., Sinclair G. I., Ruane P. J., Crofoot G. E., McDonald C. K., Brainard D. M., Lad L., Swaminathan S., Sundquist W. I., Sakowicz R., Chester A. E., Lee W. E., Daar E. S., Yant S. R., Cihlar T., Clinical targeting of HIV capsid protein with a long-acting small molecule. Nature 584, 614–618 (2020). - PMC - PubMed
-
- Collier D. A., Monit C., Gupta R. K., The impact of HIV-1 drug escape on the global treatment landscape. Cell Host Microbe 26, 48–60 (2019). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
