

Genomic evolution shapes prostate cancer disease type
- PMID: 38428419
- PMCID: PMC10943594
- DOI: 10.1016/j.xgen.2024.100511
Genomic evolution shapes prostate cancer disease type
Abstract
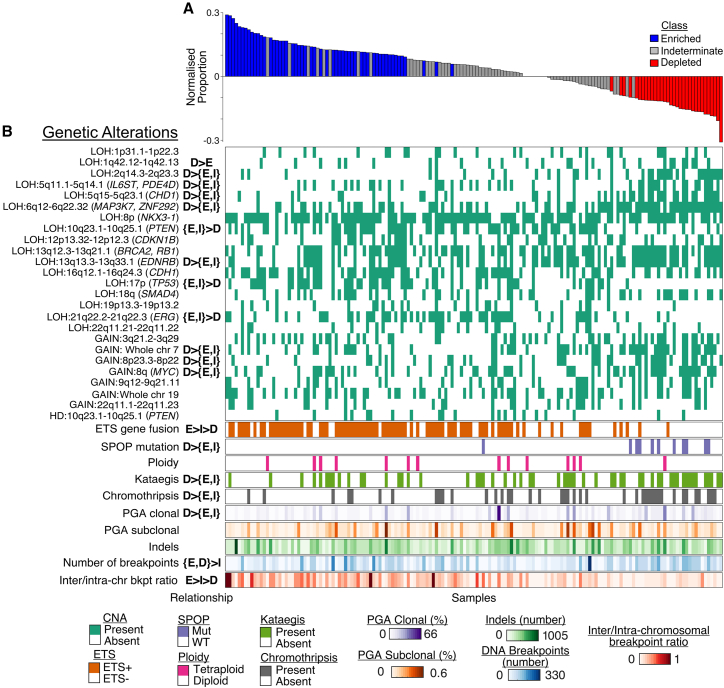
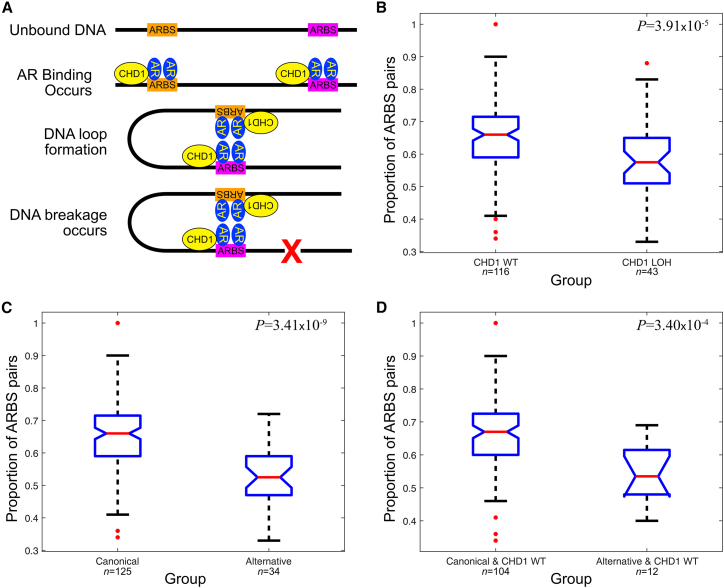
The development of cancer is an evolutionary process involving the sequential acquisition of genetic alterations that disrupt normal biological processes, enabling tumor cells to rapidly proliferate and eventually invade and metastasize to other tissues. We investigated the genomic evolution of prostate cancer through the application of three separate classification methods, each designed to investigate a different aspect of tumor evolution. Integrating the results revealed the existence of two distinct types of prostate cancer that arise from divergent evolutionary trajectories, designated as the Canonical and Alternative evolutionary disease types. We therefore propose the evotype model for prostate cancer evolution wherein Alternative-evotype tumors diverge from those of the Canonical-evotype through the stochastic accumulation of genetic alterations associated with disruptions to androgen receptor DNA binding. Our model unifies many previous molecular observations, providing a powerful new framework to investigate prostate cancer disease progression.
Keywords: AR binding; cancer evolution; evotype model; evotypes; ordering; prostate cancer.
Copyright © 2024 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests An international patent related to this work has been published under international publication no. WO 2023/047140 A1. R.A.E. has the following conflicts of interest to declare: she has received honoraria from GU-ASCO, Janssen, The University of Chicago, and Dana-Farber Cancer Institute USA as a speaker and educational honorarium from Bayer and Ipsen and is a member of the external expert committee to Astra Zeneca UK. She undertakes private practice as a sole trader at The Royal Marsden NHS Foundation Trust and 90 Sloane Street SW1X 9PQ and 280 Kings Road SW3 4NX, London, UK.
Figures






References
-
- Nowell P.C. The clonal evolution of tumor cell populations. Science. 1976;194:23–28. - PubMed
-
- Nangalia J., Nice F.L., Wedge D.C., Godfrey A.L., Grinfeld J., Thakker C., Massie C.E., Baxter J., Sewell D., Silber Y., et al. DNMT3A mutations occur early or late in patients with myeloproliferative neoplasms and mutation order influences phenotype. Haematologica. 2015;100:e438–e442. doi: 10.3324/haematol.2015.129510. - DOI - PMC - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
