

Combinatorial selective ER-phagy remodels the ER during neurogenesis
- PMID: 38429475
- PMCID: PMC10940164
- DOI: 10.1038/s41556-024-01356-4
Combinatorial selective ER-phagy remodels the ER during neurogenesis
Erratum in
-
Author Correction: Combinatorial selective ER-phagy remodels the ER during neurogenesis.Nat Cell Biol. 2025 Jun;27(6):1048. doi: 10.1038/s41556-025-01670-5. Nat Cell Biol. 2025. PMID: 40234617 Free PMC article. No abstract available.
Abstract
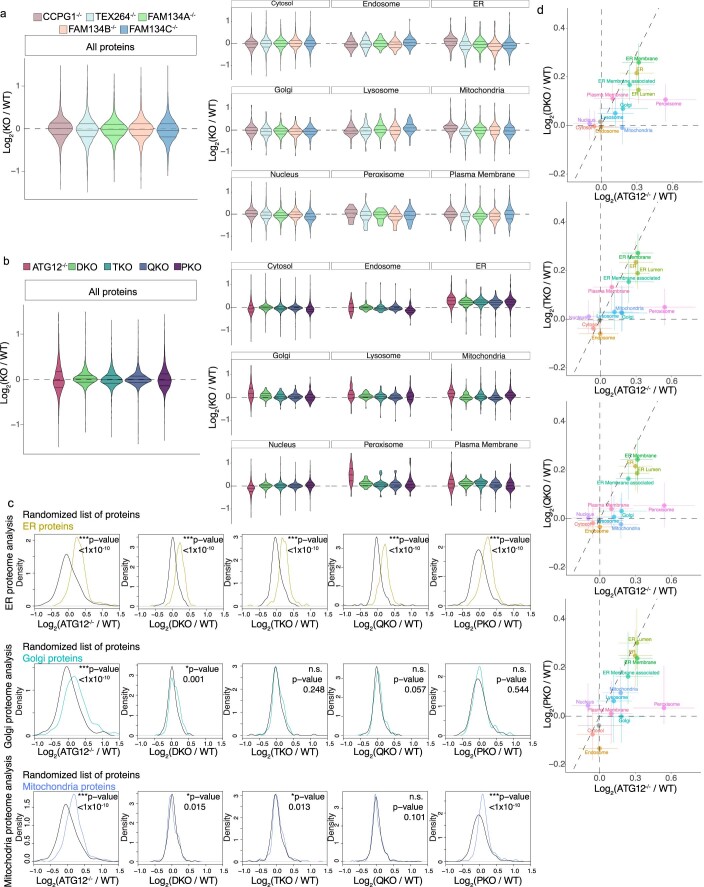
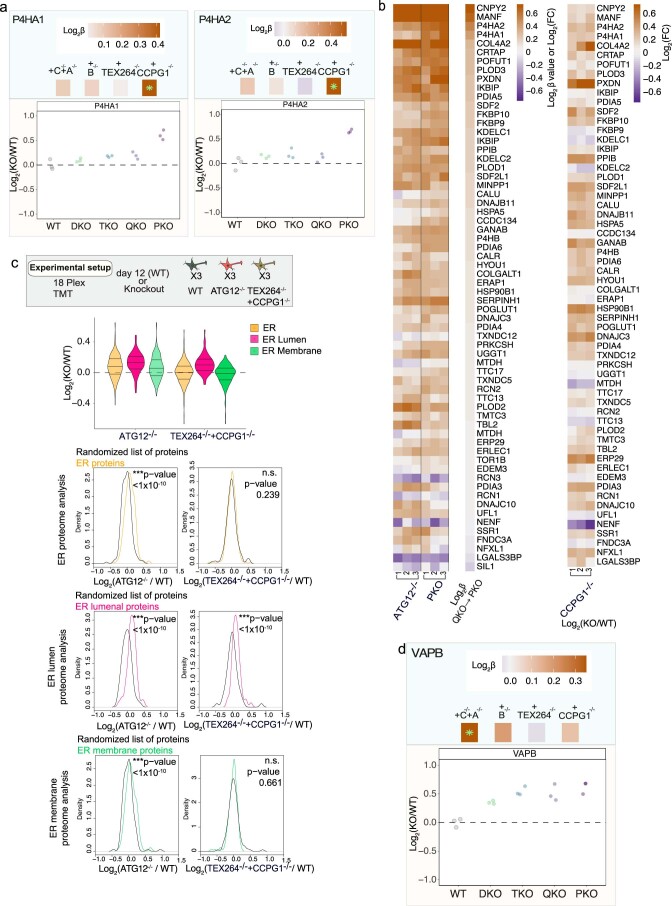
The endoplasmic reticulum (ER) employs a diverse proteome landscape to orchestrate many cellular functions, ranging from protein and lipid synthesis to calcium ion flux and inter-organelle communication. A case in point concerns the process of neurogenesis, where a refined tubular ER network is assembled via ER shaping proteins into the newly formed neuronal projections to create highly polarized dendrites and axons. Previous studies have suggested a role for autophagy in ER remodelling, as autophagy-deficient neurons in vivo display axonal ER accumulation within synaptic boutons, and the membrane-embedded ER-phagy receptor FAM134B has been genetically linked with human sensory and autonomic neuropathy. However, our understanding of the mechanisms underlying selective removal of the ER and the role of individual ER-phagy receptors is limited. Here we combine a genetically tractable induced neuron (iNeuron) system for monitoring ER remodelling during in vitro differentiation with proteomic and computational tools to create a quantitative landscape of ER proteome remodelling via selective autophagy. Through analysis of single and combinatorial ER-phagy receptor mutants, we delineate the extent to which each receptor contributes to both the magnitude and selectivity of ER protein clearance. We define specific subsets of ER membrane or lumenal proteins as preferred clients for distinct receptors. Using spatial sensors and flux reporters, we demonstrate receptor-specific autophagic capture of ER in axons, and directly visualize tubular ER membranes within autophagosomes in neuronal projections by cryo-electron tomography. This molecular inventory of ER proteome remodelling and versatile genetic toolkit provide a quantitative framework for understanding the contributions of individual ER-phagy receptors for reshaping ER during cell state transitions.
© 2024. The Author(s).
Conflict of interest statement
J.W.H. is a consultant and founder of Caraway Therapeutics (a wholly owned subsidiary of Merck & Co) and is a member of the scientific advisory board for Lyterian Therapeutics. B.A.S. is a co-founding scientific advisory board member of Interline Therapeutics and on the scientific advisory boards of Biotheryx and Proxygen. W.B. holds additional appointments as an honorary Professor at the Technical University Munich and a Distinguished Professor at ShanghaiTech University and is a member of the Life Science Advisory Board of Thermo Fisher Scientific. The other authors declare no competing interests.
Figures
















Update of
-
Combinatorial selective ER-phagy remodels the ER during neurogenesis.bioRxiv [Preprint]. 2023 Nov 10:2023.06.26.546565. doi: 10.1101/2023.06.26.546565. bioRxiv. 2023. Update in: Nat Cell Biol. 2024 Mar;26(3):378-392. doi: 10.1038/s41556-024-01356-4. PMID: 37425907 Free PMC article. Updated. Preprint.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
