

Digenic inheritance involving a muscle-specific protein kinase and the giant titin protein causes a skeletal muscle myopathy
- PMID: 38429495
- PMCID: PMC10937387
- DOI: 10.1038/s41588-023-01651-0
Digenic inheritance involving a muscle-specific protein kinase and the giant titin protein causes a skeletal muscle myopathy
Abstract
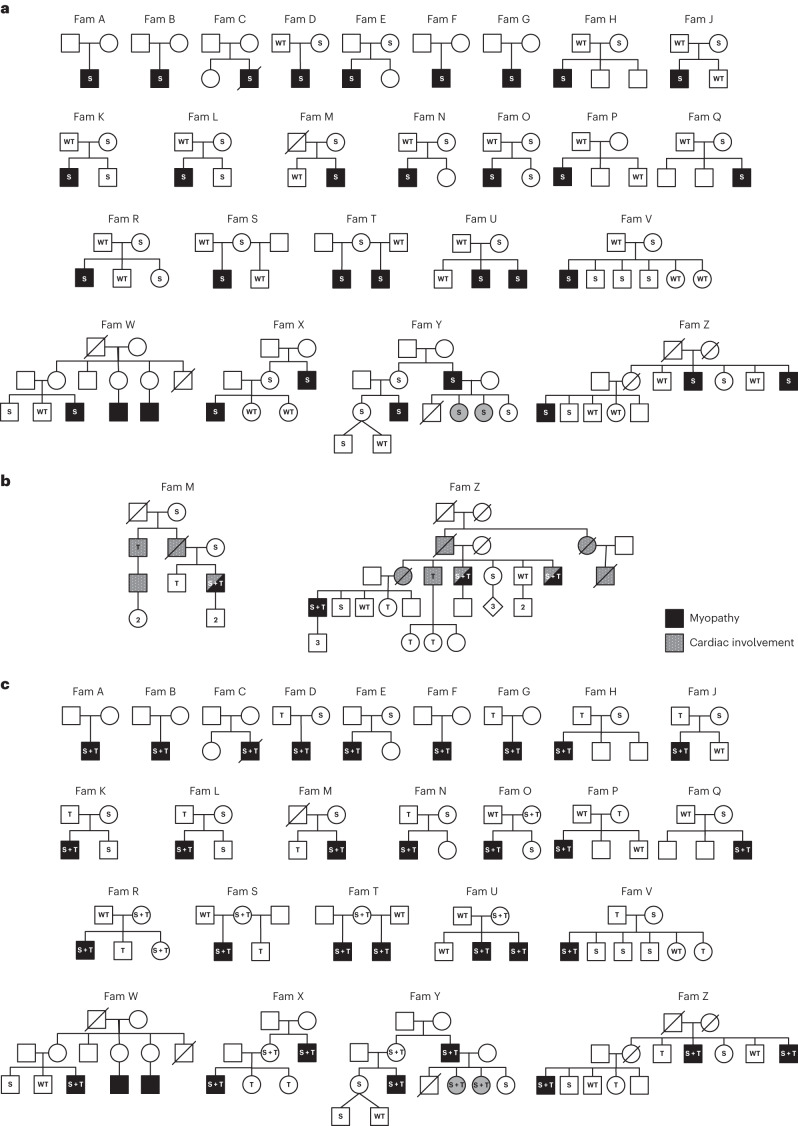
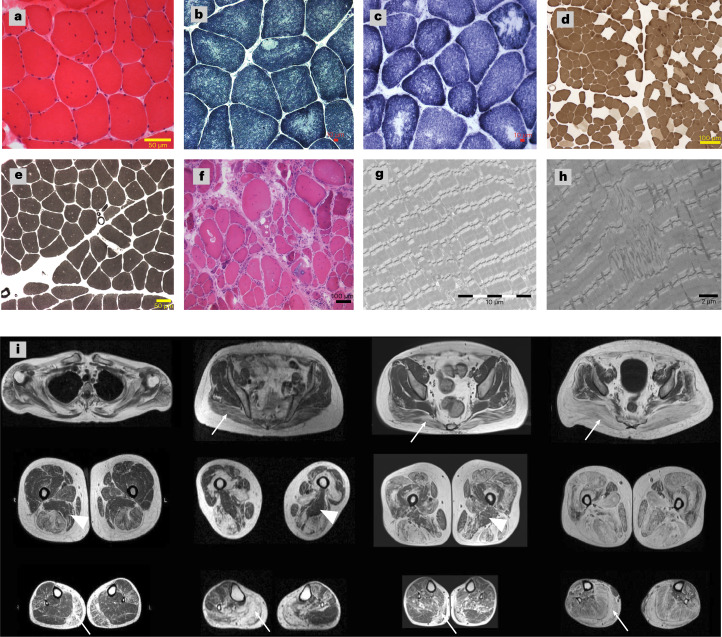
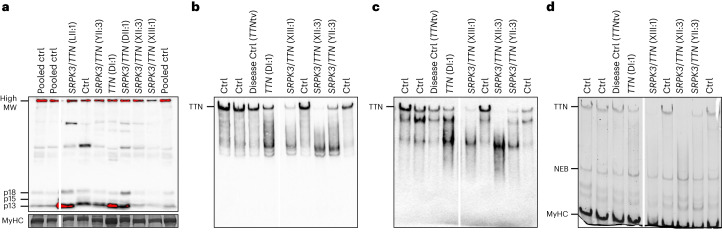
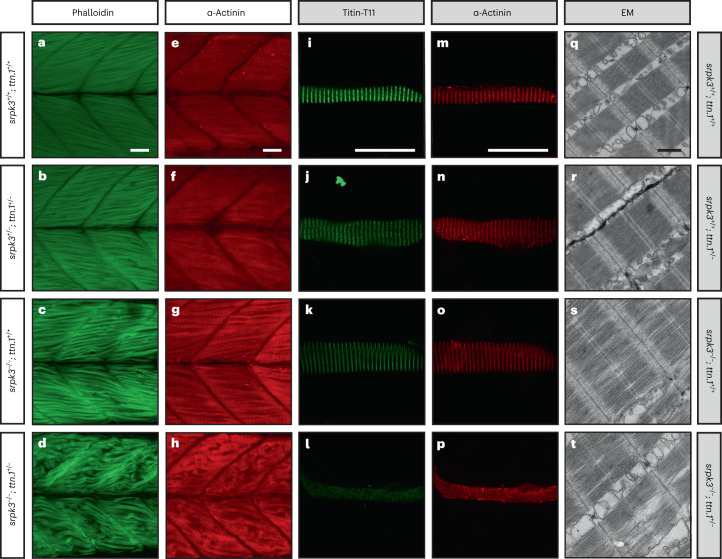
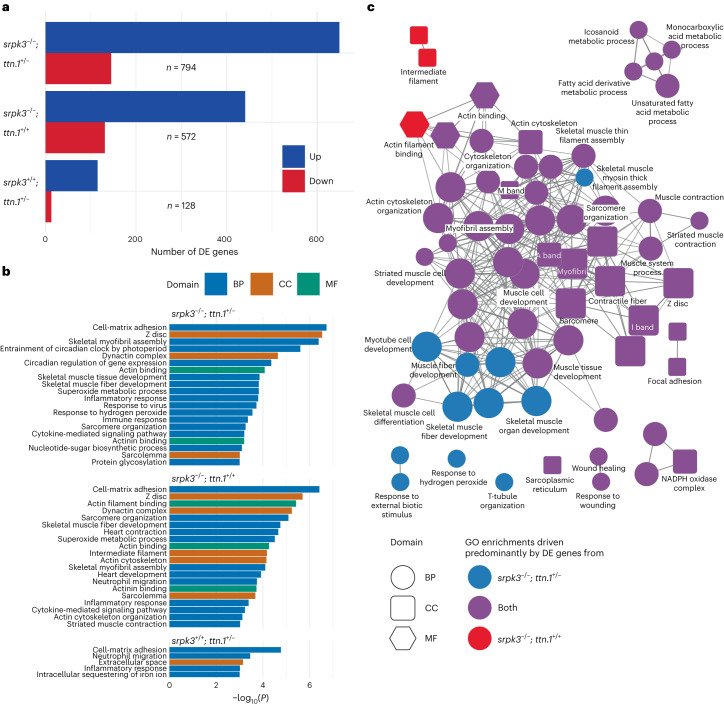
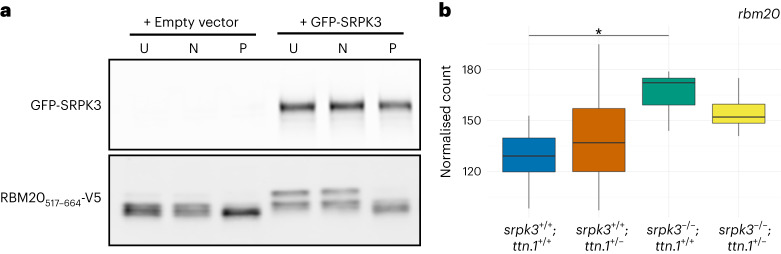
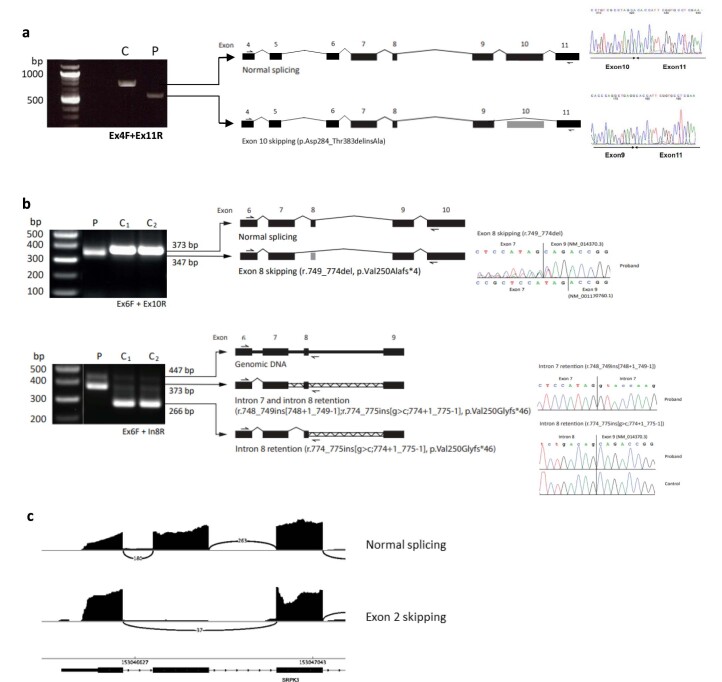
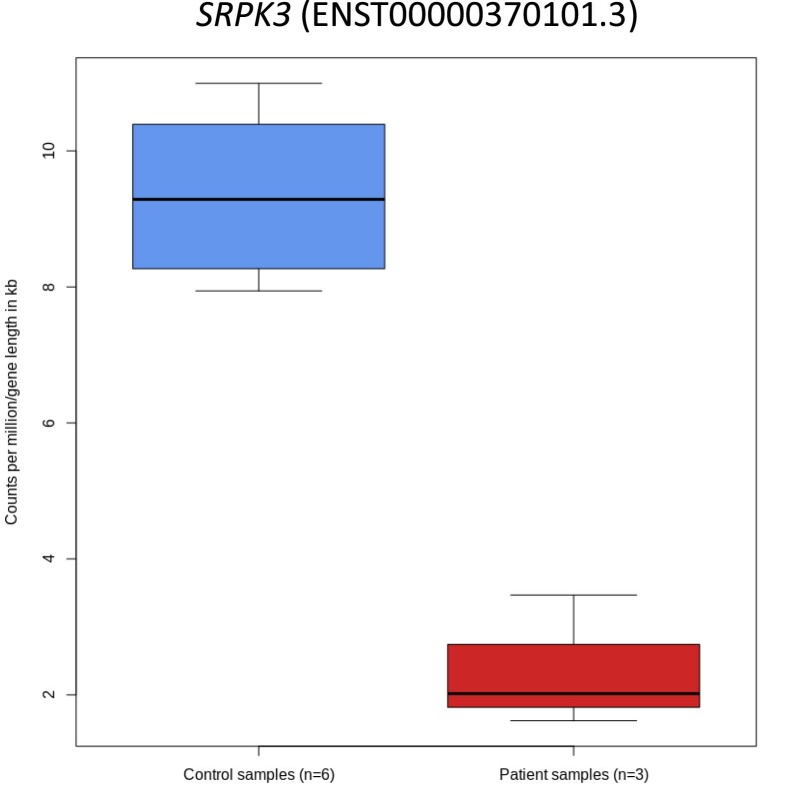
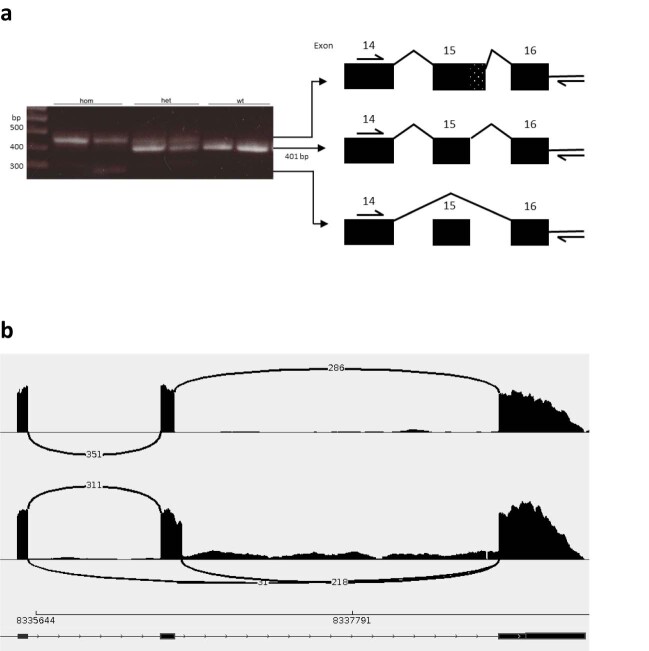
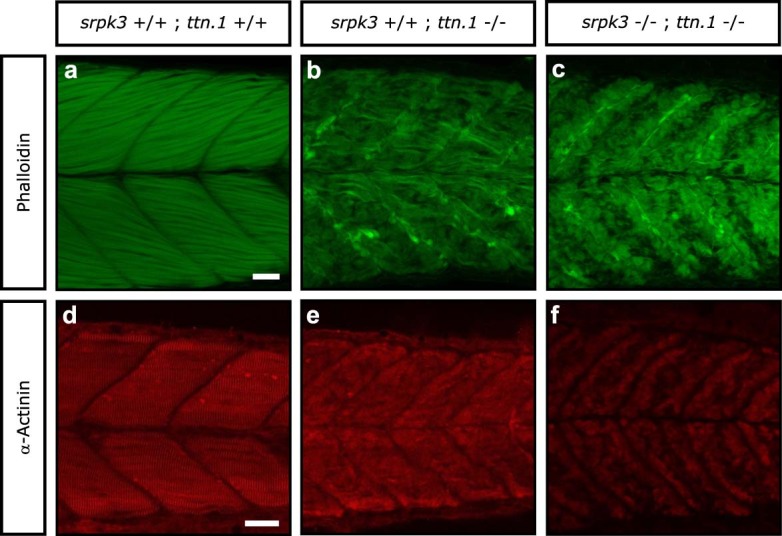
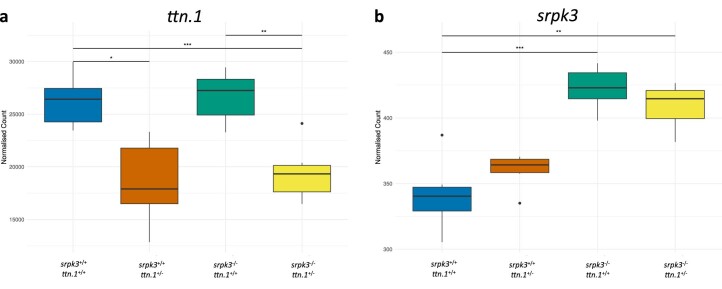
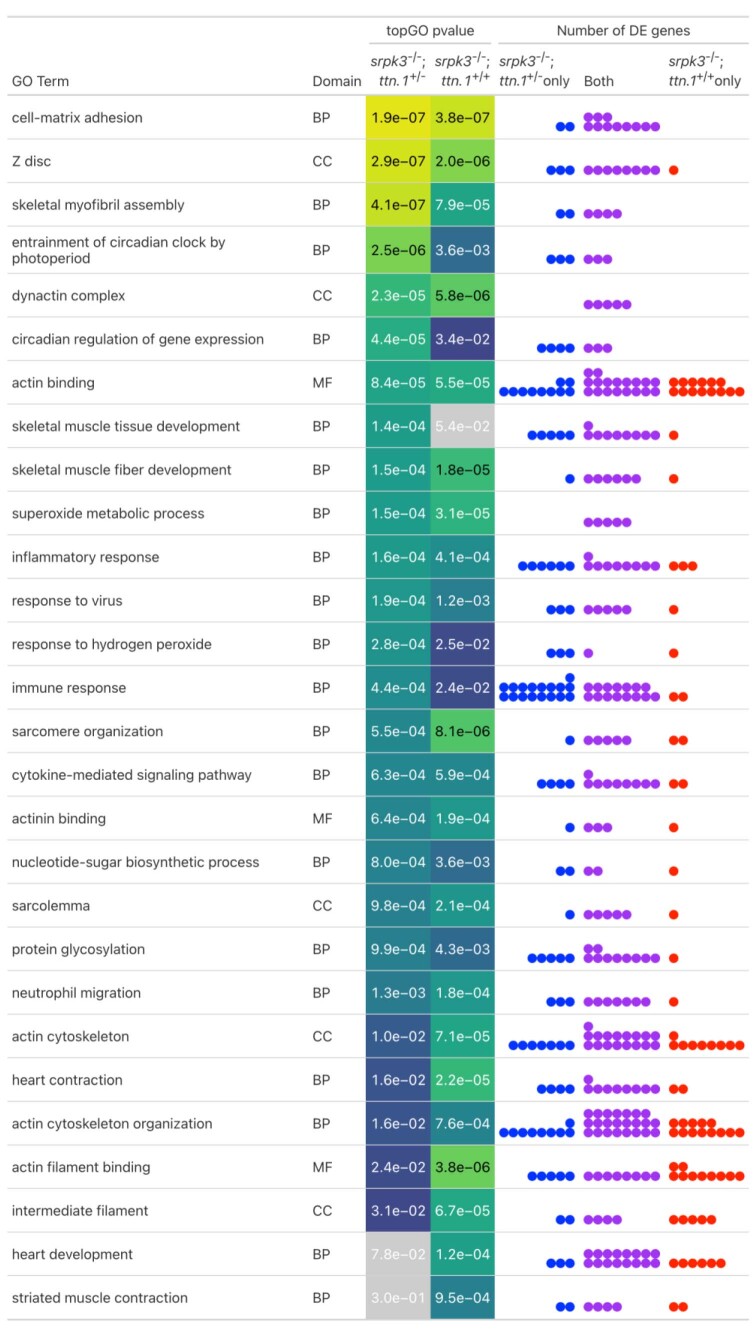
In digenic inheritance, pathogenic variants in two genes must be inherited together to cause disease. Only very few examples of digenic inheritance have been described in the neuromuscular disease field. Here we show that predicted deleterious variants in SRPK3, encoding the X-linked serine/argenine protein kinase 3, lead to a progressive early onset skeletal muscle myopathy only when in combination with heterozygous variants in the TTN gene. The co-occurrence of predicted deleterious SRPK3/TTN variants was not seen among 76,702 healthy male individuals, and statistical modeling strongly supported digenic inheritance as the best-fitting model. Furthermore, double-mutant zebrafish (srpk3-/-; ttn.1+/-) replicated the myopathic phenotype and showed myofibrillar disorganization. Transcriptome data suggest that the interaction of srpk3 and ttn.1 in zebrafish occurs at a post-transcriptional level. We propose that digenic inheritance of deleterious changes impacting both the protein kinase SRPK3 and the giant muscle protein titin causes a skeletal myopathy and might serve as a model for other genetic diseases.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
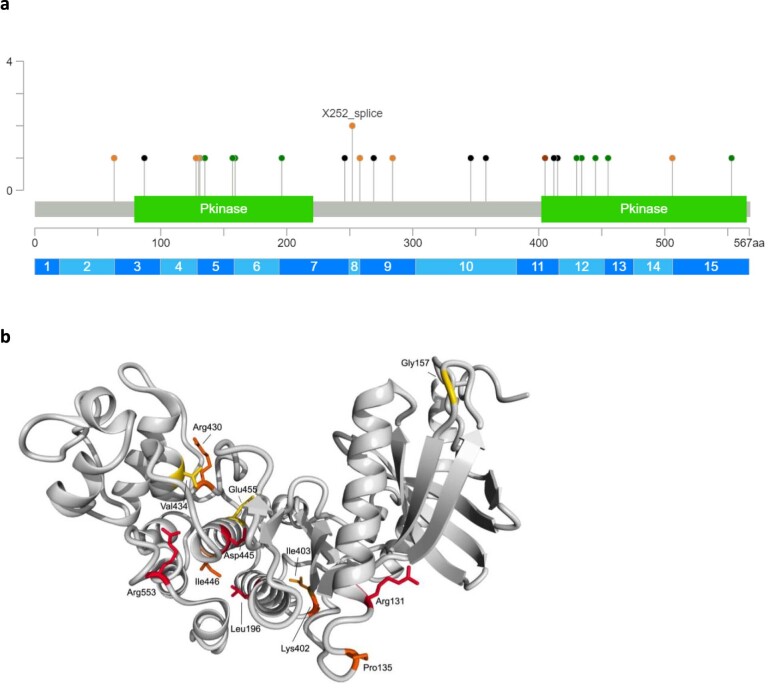
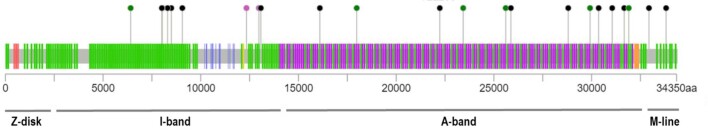
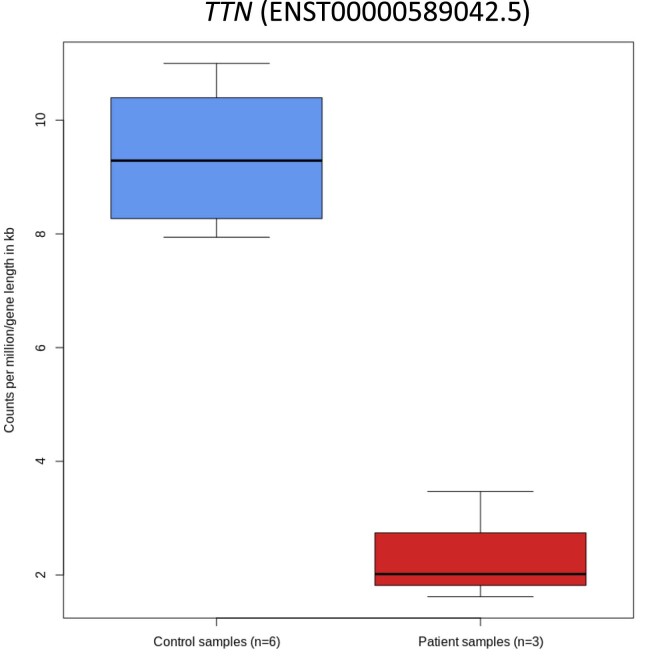
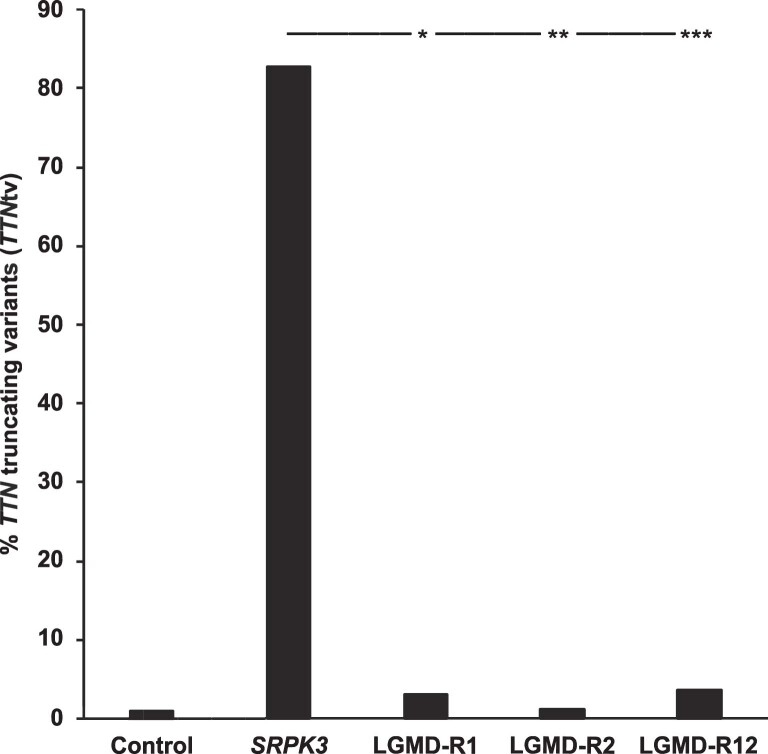
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
