

Systematic and quantitative analysis of stop codon readthrough in Rett syndrome nonsense mutations
- PMID: 38430393
- PMCID: PMC11055764
- DOI: 10.1007/s00109-024-02436-6
Systematic and quantitative analysis of stop codon readthrough in Rett syndrome nonsense mutations
Abstract
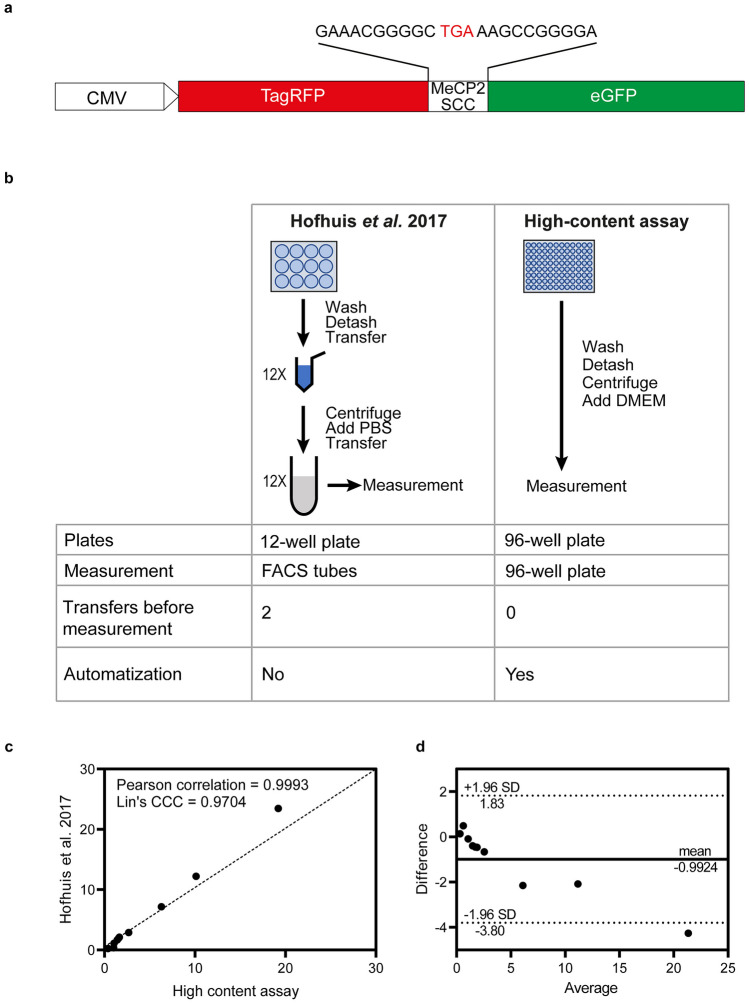
Rett syndrome (RTT) is a neurodevelopmental disorder resulting from genetic mutations in the methyl CpG binding protein 2 (MeCP2) gene. Specifically, around 35% of RTT patients harbor premature termination codons (PTCs) within the MeCP2 gene due to nonsense mutations. A promising therapeutic avenue for these individuals involves the use of aminoglycosides, which stimulate translational readthrough (TR) by causing stop codons to be interpreted as sense codons. However, the effectiveness of this treatment depends on several factors, including the type of stop codon and the surrounding nucleotides, collectively referred to as the stop codon context (SCC). Here, we develop a high-content reporter system to precisely measure TR efficiency at different SCCs, assess the recovery of the full-length MeCP2 protein, and evaluate its subcellular localization. We have conducted a comprehensive investigation into the intricate relationship between SCC characteristics and TR induction, examining a total of 14 pathogenic MeCP2 nonsense mutations with the aim to advance the prospects of personalized therapy for individuals with RTT. Our results demonstrate that TR induction can successfully restore full-length MeCP2 protein, albeit to varying degrees, contingent upon the SCC and the specific position of the PTC within the MeCP2 mRNA. TR induction can lead to the re-establishment of nuclear localization of MeCP2, indicating the potential restoration of protein functionality. In summary, our findings underscore the significance of SCC-specific approaches in the development of tailored therapies for RTT. By unraveling the relationship between SCC and TR therapy, we pave the way for personalized, individualized treatment strategies that hold promise for improving the lives of individuals affected by this debilitating neurodevelopmental disorder. KEY MESSAGES: The efficiency of readthrough induction at MeCP2 premature termination codons strongly depends on the stop codon context. The position of the premature termination codon on the transcript influences the readthrough inducibility. A new high-content dual reporter assay facilitates the measurement and prediction of readthrough efficiency of specific nucleotide stop contexts. Readthrough induction results in the recovery of full-length MeCP2 and its re-localization to the nucleus. MeCP2 requires only one of its annotated nuclear localization signals.
Keywords: MeCP2; Aminoglycoside; Personalized medicine; Rare disease; Rett syndrome; Translational readthrough.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures




Similar articles
-
Evaluation of Novel Enhancer Compounds in Gentamicin-Mediated Readthrough of Nonsense Mutations in Rett Syndrome.Int J Mol Sci. 2023 Jul 19;24(14):11665. doi: 10.3390/ijms241411665. Int J Mol Sci. 2023. PMID: 37511424 Free PMC article.
-
Ex vivo treatment with a novel synthetic aminoglycoside NB54 in primary fibroblasts from Rett syndrome patients suppresses MECP2 nonsense mutations.PLoS One. 2011;6(6):e20733. doi: 10.1371/journal.pone.0020733. Epub 2011 Jun 13. PLoS One. 2011. PMID: 21695138 Free PMC article.
-
Readthrough of nonsense mutations in Rett syndrome: evaluation of novel aminoglycosides and generation of a new mouse model.J Mol Med (Berl). 2011 Apr;89(4):389-98. doi: 10.1007/s00109-010-0704-4. Epub 2010 Dec 1. J Mol Med (Berl). 2011. PMID: 21120444 Free PMC article.
-
Translational readthrough potential of natural termination codons in eucaryotes--The impact of RNA sequence.RNA Biol. 2015;12(9):950-8. doi: 10.1080/15476286.2015.1068497. RNA Biol. 2015. PMID: 26176195 Free PMC article. Review.
-
Factors Affecting Readthrough of Natural Versus Premature Termination Codons.Adv Exp Med Biol. 2023;1415:149-155. doi: 10.1007/978-3-031-27681-1_23. Adv Exp Med Biol. 2023. PMID: 37440028 Review.
Cited by
-
Defining the high-translational readthrough stop codon context.PLoS Genet. 2025 Jun 25;21(6):e1011753. doi: 10.1371/journal.pgen.1011753. eCollection 2025 Jun. PLoS Genet. 2025. PMID: 40561183 Free PMC article.
-
When "loss-of-function" means proteostasis burden: Thinking again about coding DNA variants.Am J Hum Genet. 2025 Jan 2;112(1):3-10. doi: 10.1016/j.ajhg.2024.12.002. Am J Hum Genet. 2025. PMID: 39753117 Free PMC article.
-
Codon Usage Evolution in Viruses: Implications for Survival and Pathogenicity.J Mol Evol. 2025 Sep 4. doi: 10.1007/s00239-025-10263-7. Online ahead of print. J Mol Evol. 2025. PMID: 40906273 Review.
References
-
- Cuddapah VA, Pillai RB, Shekar KV, Lane JB, Motil KJ, Skinner SA, Tarquinio DC, Glaze DG, McGwin G, Kaufmann WE, Percy AK, Neul JL, Olsen ML. Methyl-CpG-binding protein 2 (MEPC2) mutation type is associated with disease severity in Rett Syndrome. J Med Genet. 2014;51:152–158. doi: 10.1136/jmedgenet-2013-102113. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
