The transcription factor NF-κB orchestrates nucleosome remodeling during the primary response to Toll-like receptor 4 signaling
- PMID: 38430908
- PMCID: PMC10984581
- DOI: 10.1016/j.immuni.2024.02.004
The transcription factor NF-κB orchestrates nucleosome remodeling during the primary response to Toll-like receptor 4 signaling
Abstract
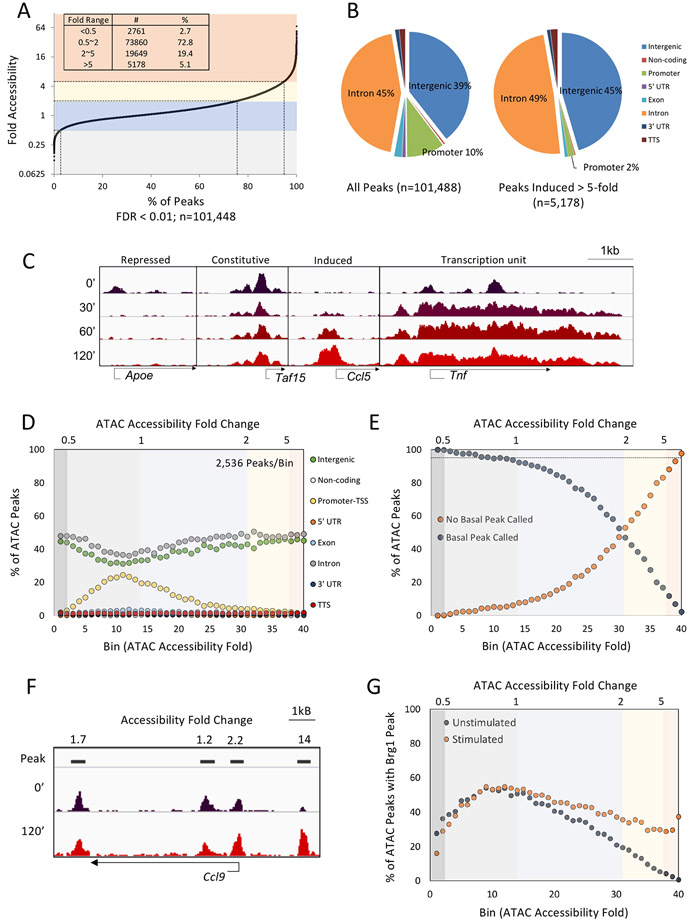
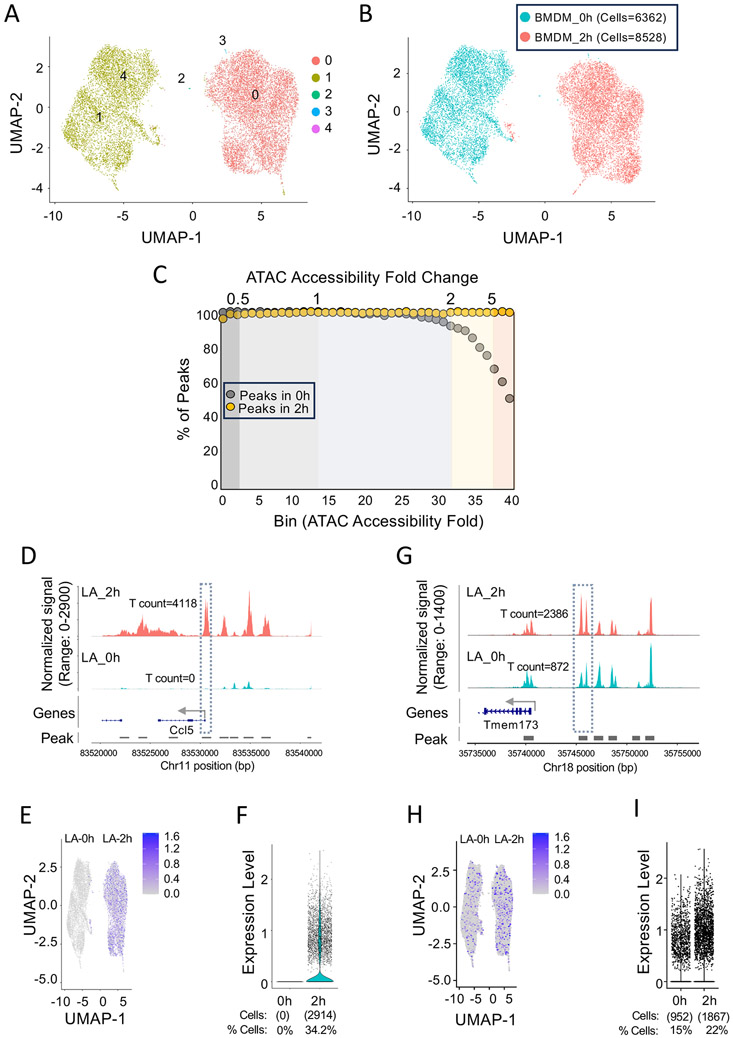
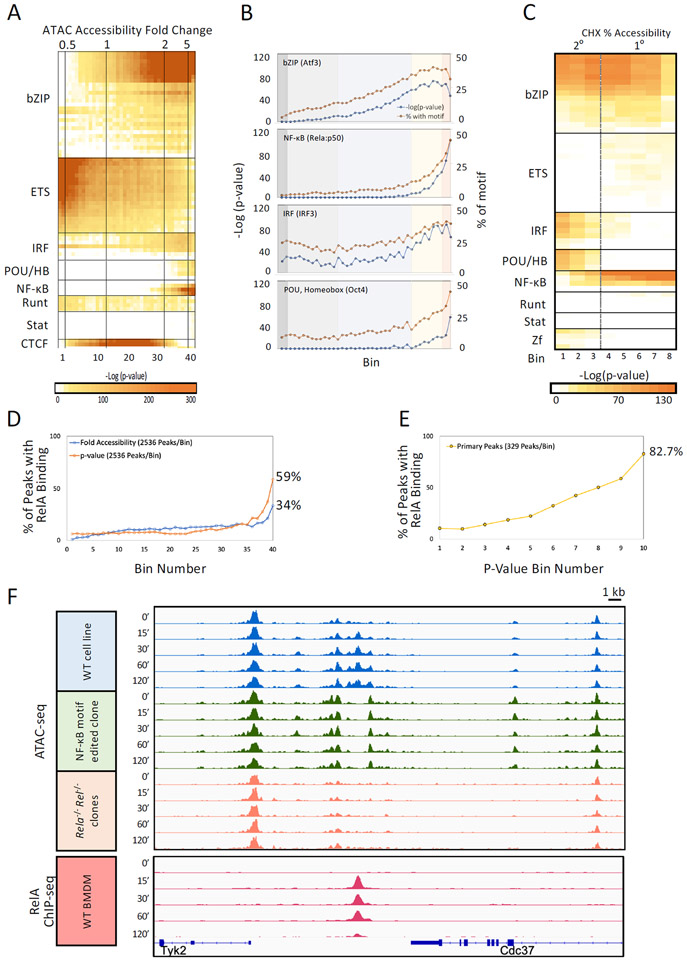
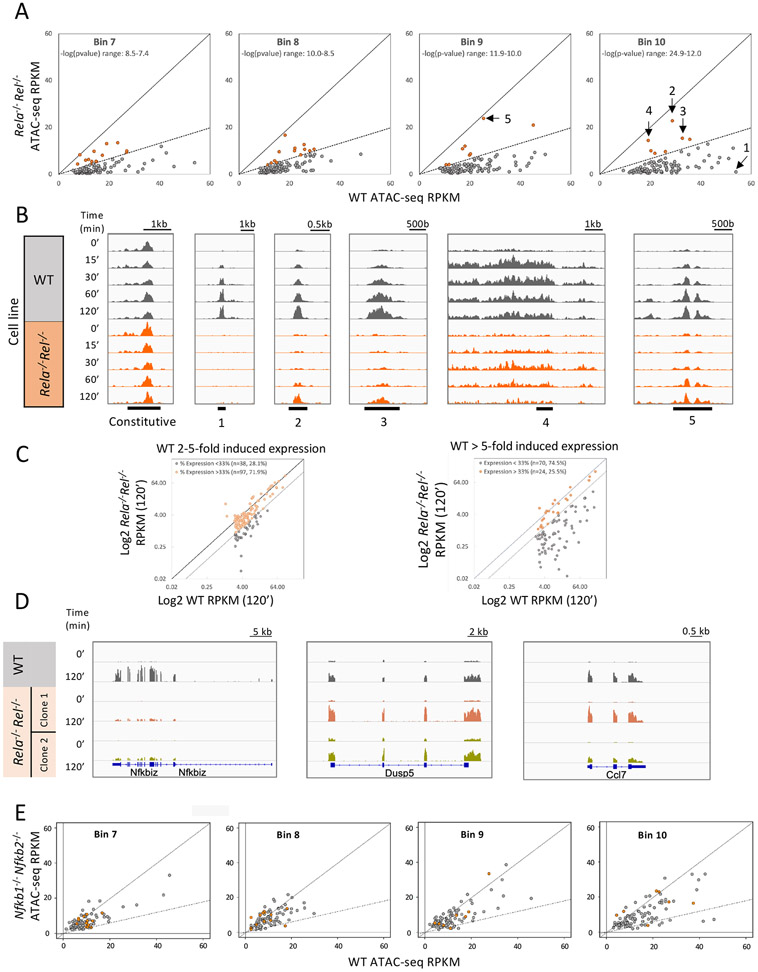
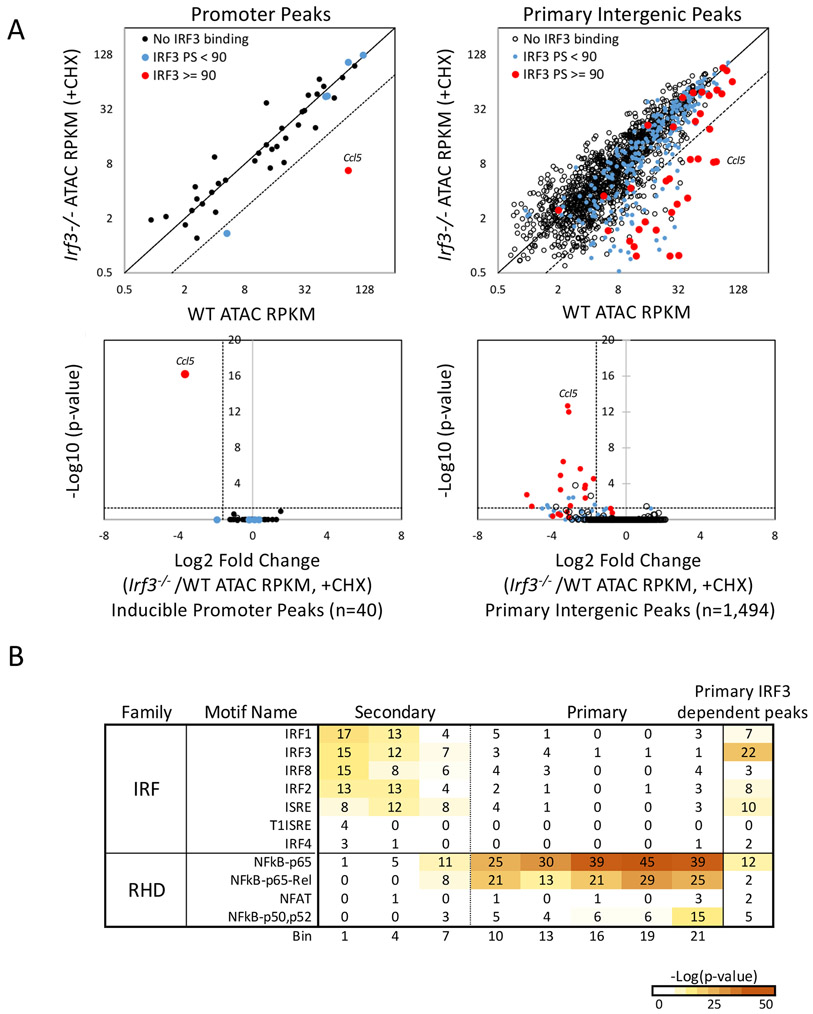
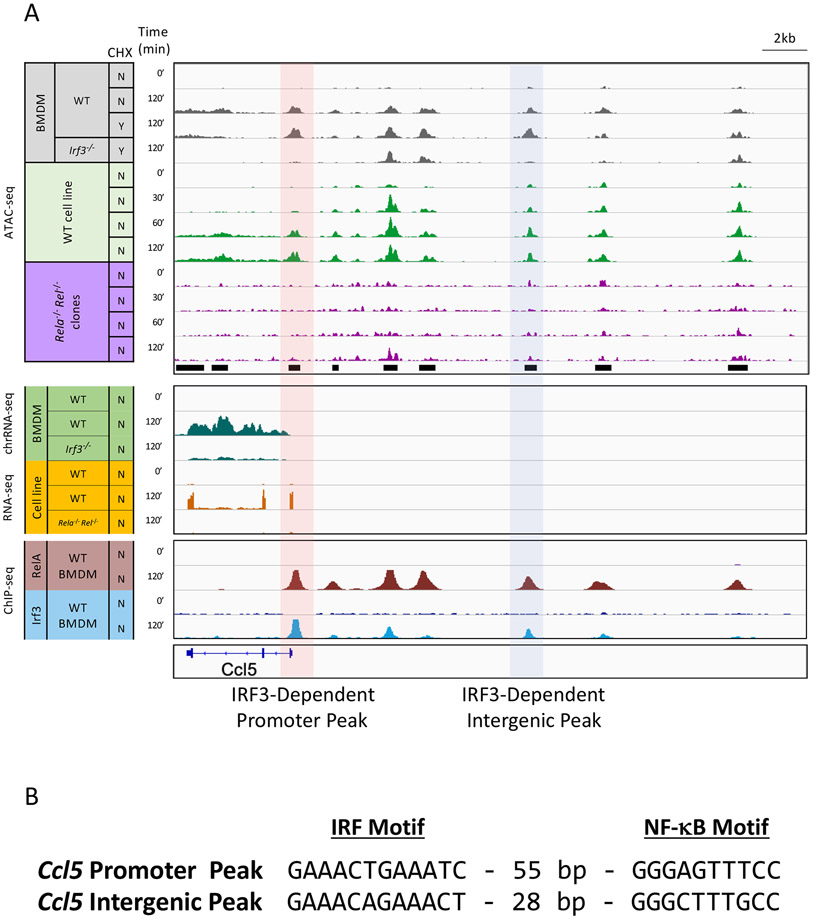
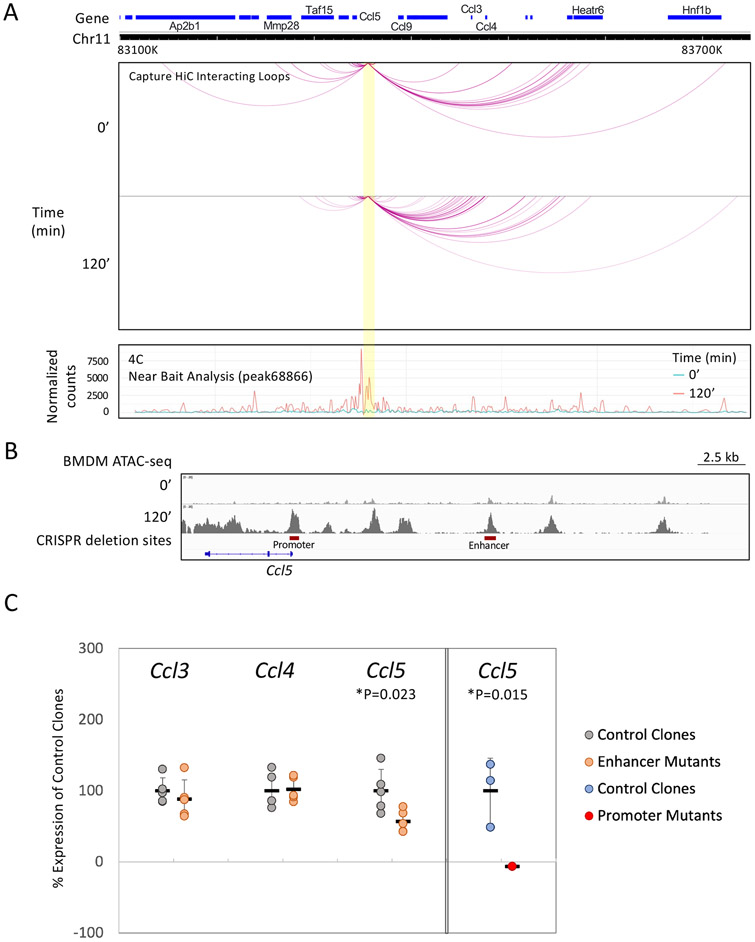
Inducible nucleosome remodeling at hundreds of latent enhancers and several promoters shapes the transcriptional response to Toll-like receptor 4 (TLR4) signaling in macrophages. We aimed to define the identities of the transcription factors that promote TLR-induced remodeling. An analysis strategy based on ATAC-seq and single-cell ATAC-seq that enriched for genomic regions most likely to undergo remodeling revealed that the transcription factor nuclear factor κB (NF-κB) bound to all high-confidence peaks marking remodeling during the primary response to the TLR4 ligand, lipid A. Deletion of NF-κB subunits RelA and c-Rel resulted in the loss of remodeling at high-confidence ATAC-seq peaks, and CRISPR-Cas9 mutagenesis of NF-κB-binding motifs impaired remodeling. Remodeling selectivity at defined regions was conferred by collaboration with other inducible factors, including IRF3- and MAP-kinase-induced factors. Thus, NF-κB is unique among TLR4-activated transcription factors in its broad contribution to inducible nucleosome remodeling, alongside its ability to activate poised enhancers and promoters assembled into open chromatin.
Keywords: IRF3; NF-κB; chromatin; macrophages; nucleosome remodeling; transcription.
Copyright © 2024 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures