

Quantitative pathogenicity and host adaptation in a fungal plant pathogen revealed by whole-genome sequencing
- PMID: 38431601
- PMCID: PMC10908820
- DOI: 10.1038/s41467-024-46191-1
Quantitative pathogenicity and host adaptation in a fungal plant pathogen revealed by whole-genome sequencing
Abstract
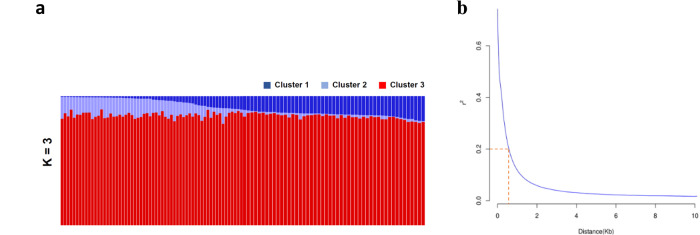
Knowledge of genetic determinism and evolutionary dynamics mediating host-pathogen interactions is essential to manage fungal plant diseases. Studies on the genetic architecture of fungal pathogenicity often focus on large-effect effector genes triggering strong, qualitative resistance. It is not clear how this translates to predominately quantitative interactions. Here, we use the Zymoseptoria tritici-wheat model to elucidate the genetic architecture of quantitative pathogenicity and mechanisms mediating host adaptation. With a multi-host genome-wide association study, we identify 19 high-confidence candidate genes associated with quantitative pathogenicity. Analysis of genetic diversity reveals that sequence polymorphism is the main evolutionary process mediating differences in quantitative pathogenicity, a process that is likely facilitated by genetic recombination and transposable element dynamics. Finally, we use functional approaches to confirm the role of an effector-like gene and a methyltransferase in phenotypic variation. This study highlights the complex genetic architecture of quantitative pathogenicity, extensive diversifying selection and plausible mechanisms facilitating pathogen adaptation.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures





References
MeSH terms
Grants and funding
- ANR-12-ADAP-0009-04/Agence Nationale de la Recherche (French National Research Agency)
- ANR-17-EUR-0007/Agence Nationale de la Recherche (French National Research Agency)
- ANR-17-EUR-0007/Agence Nationale de la Recherche (French National Research Agency)
- ANR-12-ADAP-0009-04/Agence Nationale de la Recherche (French National Research Agency)
- ANR-12-ADAP-0009-04/Agence Nationale de la Recherche (French National Research Agency)
LinkOut - more resources
Full Text Sources
