

From germline genome to highly fragmented somatic genome: genome-wide DNA rearrangement during the sexual process in ciliated protists
- PMID: 38433968
- PMCID: PMC10901763
- DOI: 10.1007/s42995-023-00213-x
From germline genome to highly fragmented somatic genome: genome-wide DNA rearrangement during the sexual process in ciliated protists
Abstract
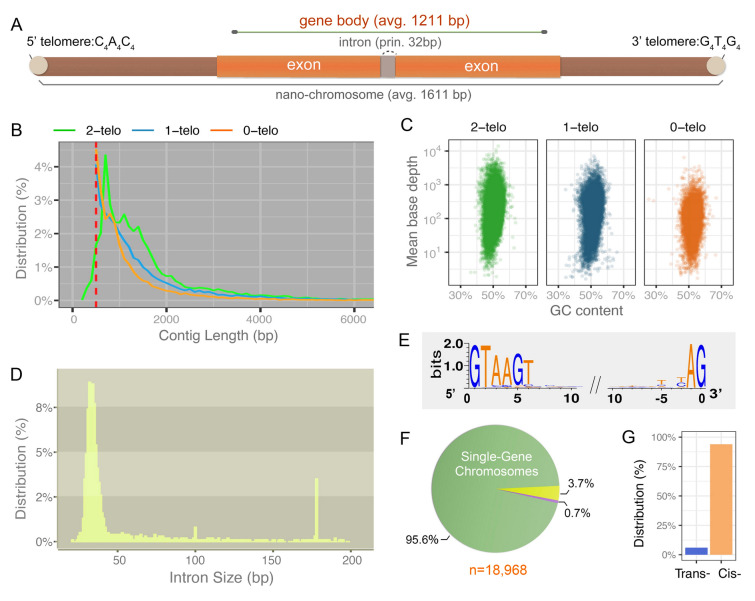
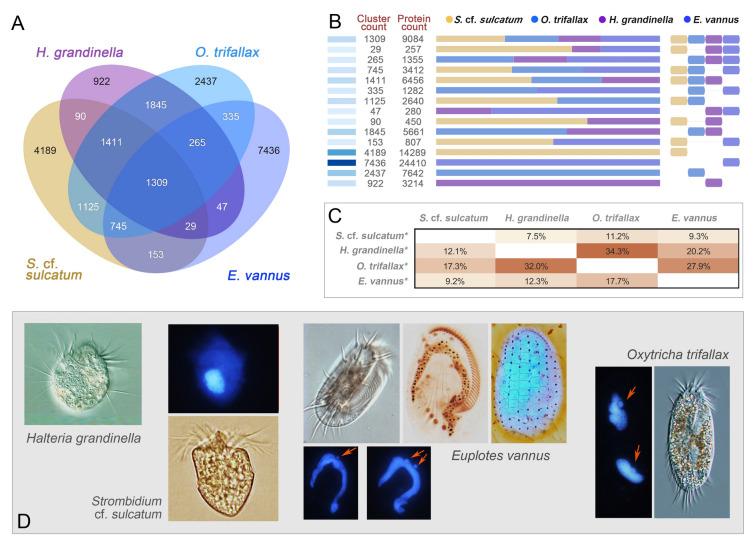
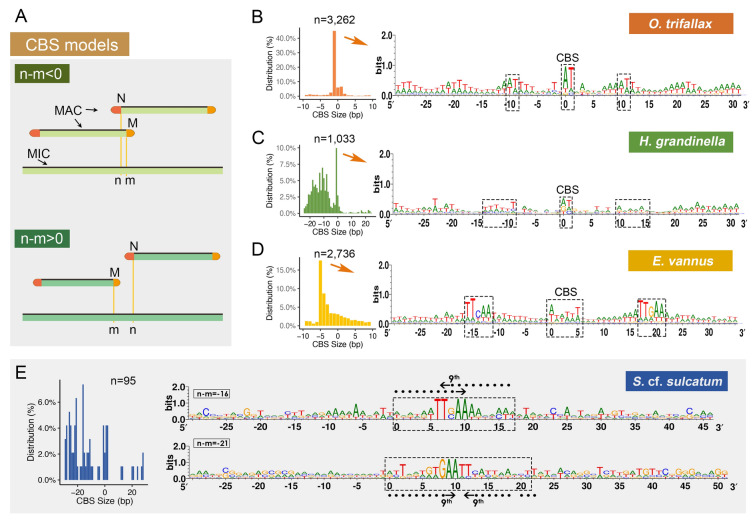
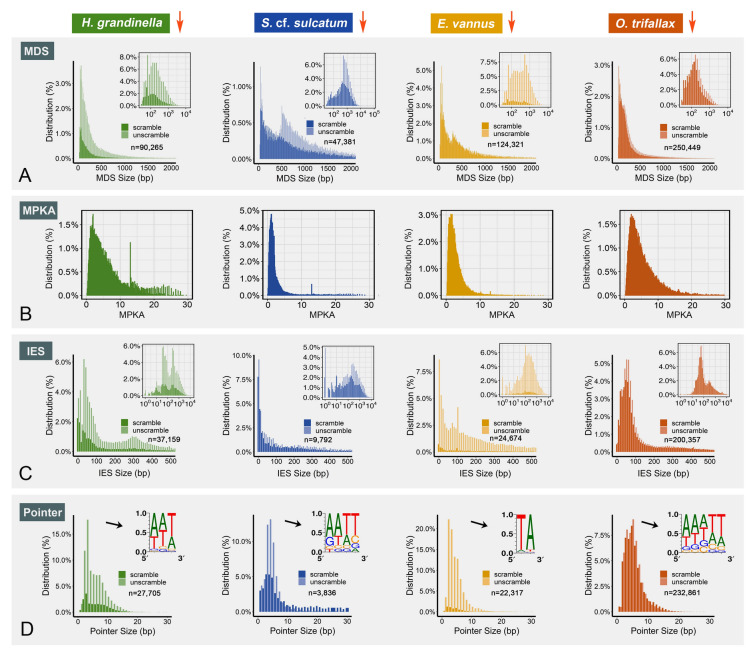
Genomes are incredibly dynamic within diverse eukaryotes and programmed genome rearrangements (PGR) play important roles in generating genomic diversity. However, genomes and chromosomes in metazoans are usually large in size which prevents our understanding of the origin and evolution of PGR. To expand our knowledge of genomic diversity and the evolutionary origin of complex genome rearrangements, we focus on ciliated protists (ciliates). Ciliates are single-celled eukaryotes with highly fragmented somatic chromosomes and massively scrambled germline genomes. PGR in ciliates occurs extensively by removing massive amounts of repetitive and selfish DNA elements found in the silent germline genome during development of the somatic genome. We report the partial germline genomes of two spirotrich ciliate species, namely Strombidium cf. sulcatum and Halteria grandinella, along with the most compact and highly fragmented somatic genome for S. cf. sulcatum. We provide the first insights into the genome rearrangements of these two species and compare these features with those of other ciliates. Our analyses reveal: (1) DNA sequence loss through evolution and during PGR in S. cf. sulcatum has combined to produce the most compact and efficient nanochromosomes observed to date; (2) the compact, transcriptome-like somatic genome in both species results from extensive removal of a relatively large number of shorter germline-specific DNA sequences; (3) long chromosome breakage site motifs are duplicated and retained in the somatic genome, revealing a complex model of chromosome fragmentation in spirotrichs; (4) gene scrambling and alternative processing are found throughout the core spirotrichs, offering unique opportunities to increase genetic diversity and regulation in this group.
Supplementary information: The online version contains supplementary material available at 10.1007/s42995-023-00213-x.
Keywords: Alternative processing; Ciliates; Gene scrambling; Genome rearrangement; Germline genome; Somatic genome.
© The Author(s) 2024.
Conflict of interest statement
Conflict of interestAll the authors declare that there are no conflicts of interest.
Figures


Similar articles
-
Exploration of the Germline Genome of the Ciliate Chilodonella uncinata through Single-Cell Omics (Transcriptomics and Genomics).mBio. 2018 Jan 9;9(1):e01836-17. doi: 10.1128/mBio.01836-17. mBio. 2018. PMID: 29317511 Free PMC article.
-
Analyses of alternatively processed genes in ciliates provide insights into the origins of scrambled genomes and may provide a mechanism for speciation.mBio. 2015 Feb 3;6(1):e01998-14. doi: 10.1128/mBio.01998-14. mBio. 2015. PMID: 25650397 Free PMC article.
-
Comparative genomics reveals insight into the evolutionary origin of massively scrambled genomes.Elife. 2022 Nov 24;11:e82979. doi: 10.7554/eLife.82979. Elife. 2022. PMID: 36421078 Free PMC article.
-
Transposon domestication versus mutualism in ciliate genome rearrangements.PLoS Genet. 2013;9(8):e1003659. doi: 10.1371/journal.pgen.1003659. Epub 2013 Aug 1. PLoS Genet. 2013. PMID: 23935529 Free PMC article. Review.
-
Macronuclear development in ciliates, with a focus on nuclear architecture.J Eukaryot Microbiol. 2022 Sep;69(5):e12898. doi: 10.1111/jeu.12898. Epub 2022 Mar 16. J Eukaryot Microbiol. 2022. PMID: 35178799 Free PMC article. Review.
Cited by
-
Novel findings on the mitochondria in ciliates, with description of mitochondrial genomes of six representatives.Mar Life Sci Technol. 2024 Sep 23;7(1):79-95. doi: 10.1007/s42995-024-00249-7. eCollection 2025 Feb. Mar Life Sci Technol. 2024. PMID: 40027321 Free PMC article.
-
Genome content reorganization in the non-model ciliate Chilodonella uncinata: insights into nuclear architecture, DNA content, and chromosome fragmentation during macronuclear development.mSphere. 2025 Jun 25;10(6):e0007525. doi: 10.1128/msphere.00075-25. Epub 2025 May 9. mSphere. 2025. PMID: 40340440 Free PMC article.
-
Dual modes of DNA N6-methyladenine maintenance by distinct methyltransferase complexes.Proc Natl Acad Sci U S A. 2025 Jan 21;122(3):e2413037121. doi: 10.1073/pnas.2413037121. Epub 2025 Jan 15. Proc Natl Acad Sci U S A. 2025. PMID: 39813249 Free PMC article.
-
Comprehensive genome annotation of the model ciliate Tetrahymena thermophila by in-depth epigenetic and transcriptomic profiling.Nucleic Acids Res. 2025 Jan 11;53(2):gkae1177. doi: 10.1093/nar/gkae1177. Nucleic Acids Res. 2025. PMID: 39657783 Free PMC article.
-
Application of RNA interference and protein localization to investigate housekeeping and developmentally regulated genes in the emerging model protozoan Paramecium caudatum.Commun Biol. 2024 Feb 19;7(1):204. doi: 10.1038/s42003-024-05906-2. Commun Biol. 2024. PMID: 38374195 Free PMC article.
References
-
- Arnaiz O, Mathy N, Baudry C, Malinsky S, Aury J-M, Wilkes CD, Garnier O, Labadie K, Lauderdale BE, Le Mouël A, Marmignon A, Nowacki M, Poulain J, Prajer M, Wincker P, Meyer E, Duharcourt S, Duret L, Bétermier M, Sperling L. The Paramecium germline genome provides a niche for intragenic parasitic DNA: evolutionary dynamics of internal eliminated sequences. PLoS Genet. 2012;8:e1002984. doi: 10.1371/journal.pgen.1002984. - DOI - PMC - PubMed
-
- Aury J-M, Jaillon O, Duret L, Noel B, Jubin C, Porcel BM, Ségurens B, Daubin V, Anthouard V, Aiach N, Arnaiz O, Billaut A, Beisson J, Blanc I, Bouhouche K, Câmara F, Duharcourt S, Guigo R, Gogendeau D, Katinka M, et al. Global trends of whole-genome duplications revealed by the ciliate Paramecium tetraurelia. Nature. 2006;444:171–178. doi: 10.1038/nature05230. - DOI - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
