

QTL mapping of human retina DNA methylation identifies 87 gene-epigenome interactions in age-related macular degeneration
- PMID: 38438351
- PMCID: PMC10912779
- DOI: 10.1038/s41467-024-46063-8
QTL mapping of human retina DNA methylation identifies 87 gene-epigenome interactions in age-related macular degeneration
Abstract
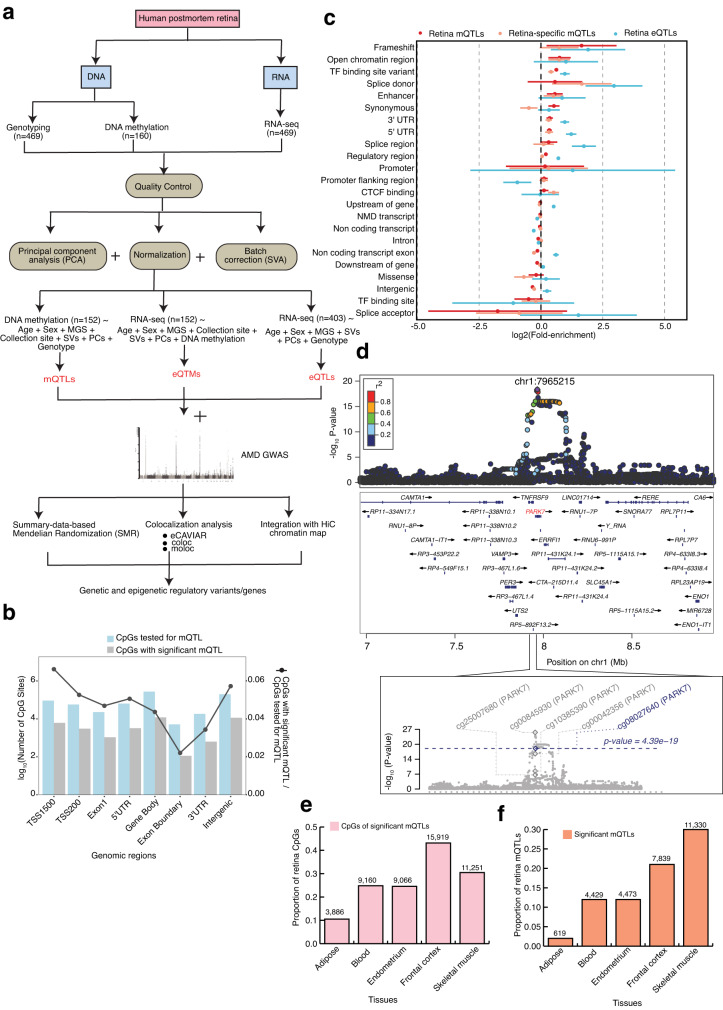
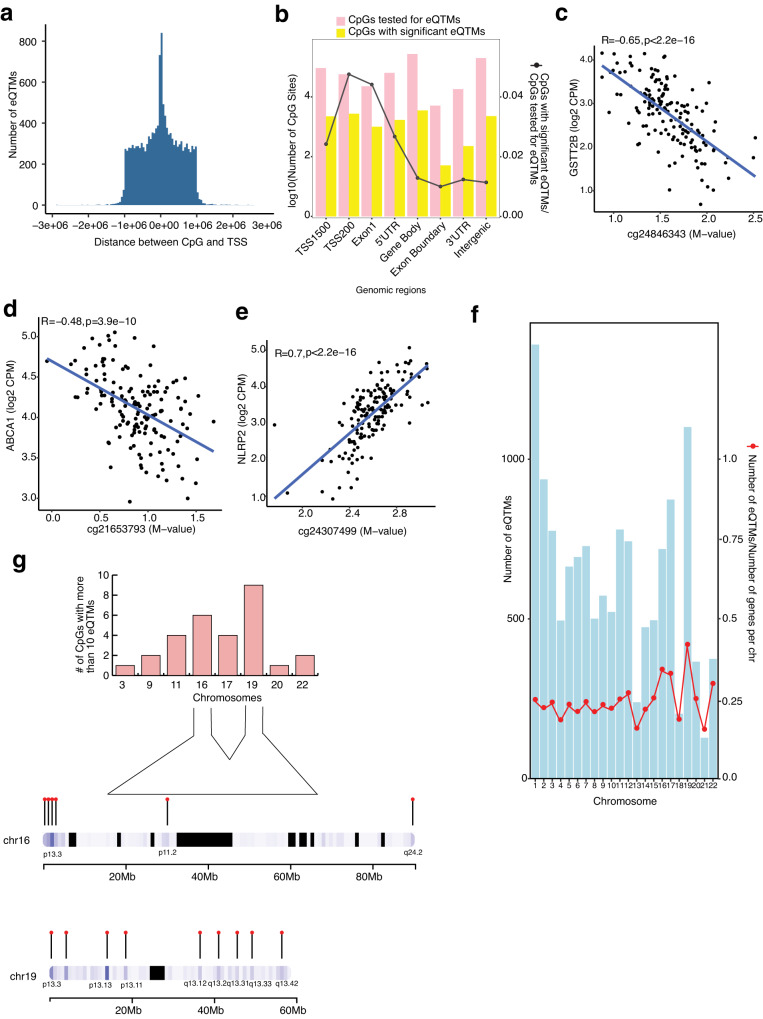
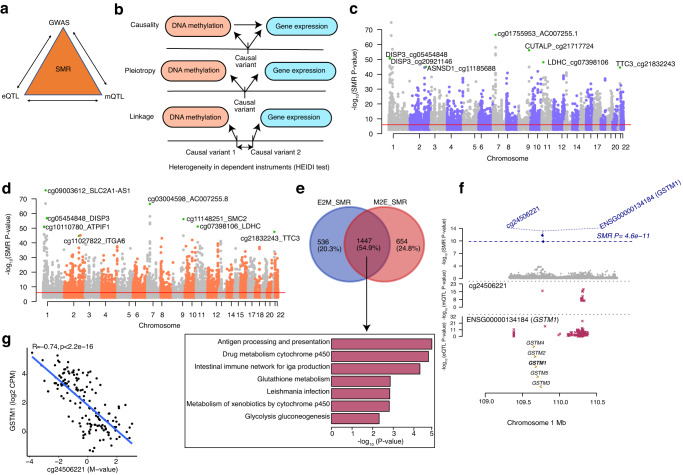
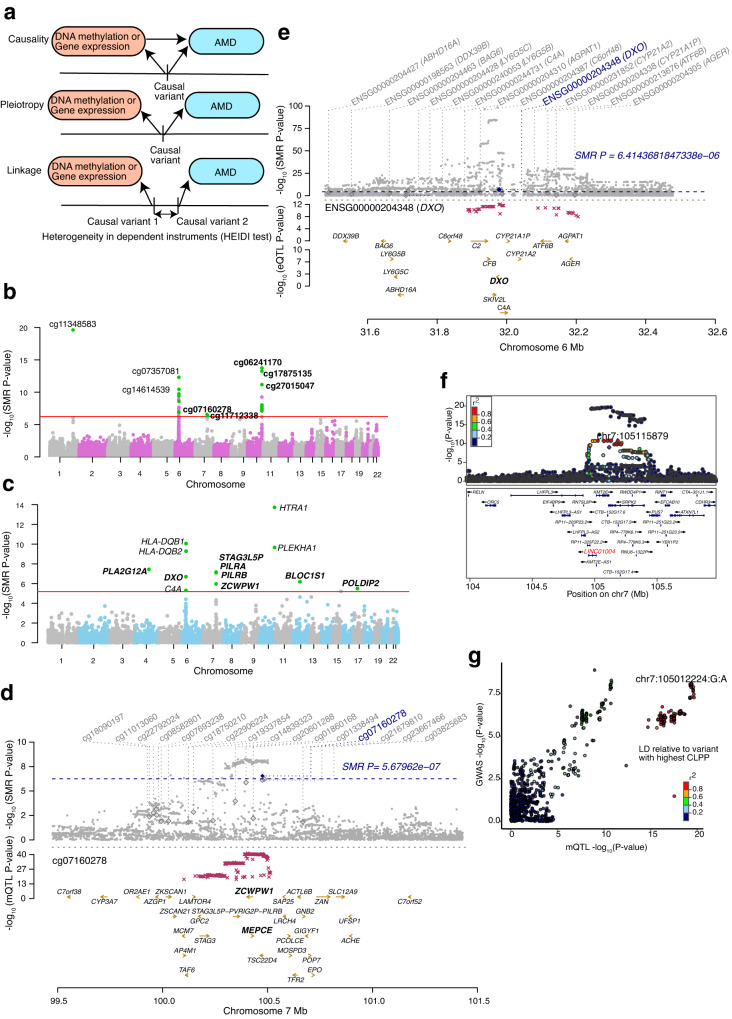
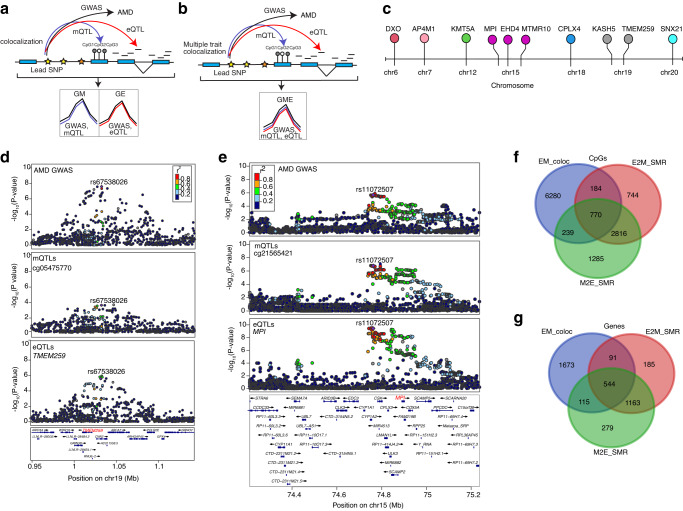
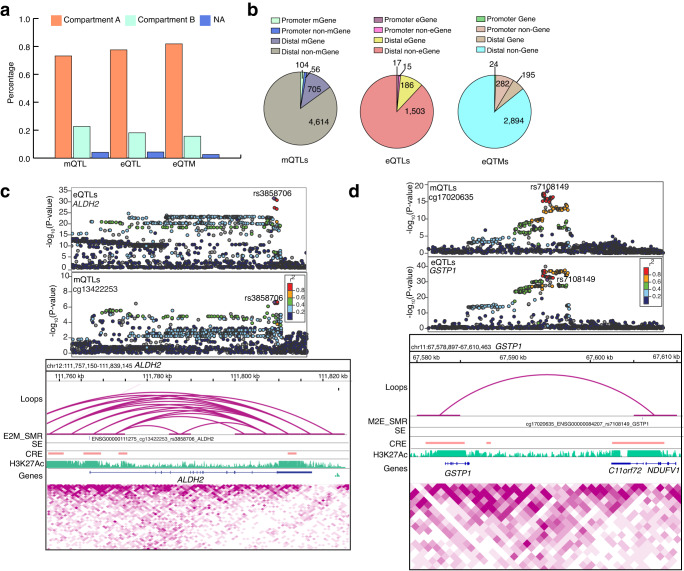
DNA methylation provides a crucial epigenetic mark linking genetic variations to environmental influence. We have analyzed array-based DNA methylation profiles of 160 human retinas with co-measured RNA-seq and >8 million genetic variants, uncovering sites of genetic regulation in cis (37,453 methylation quantitative trait loci and 12,505 expression quantitative trait loci) and 13,747 DNA methylation loci affecting gene expression, with over one-third specific to the retina. Methylation and expression quantitative trait loci show non-random distribution and enrichment of biological processes related to synapse, mitochondria, and catabolism. Summary data-based Mendelian randomization and colocalization analyses identify 87 target genes where methylation and gene-expression changes likely mediate the genotype effect on age-related macular degeneration. Integrated pathway analysis reveals epigenetic regulation of immune response and metabolism including the glutathione pathway and glycolysis. Our study thus defines key roles of genetic variations driving methylation changes, prioritizes epigenetic control of gene expression, and suggests frameworks for regulation of macular degeneration pathology by genotype-environment interaction in retina.
© 2024. This is a U.S. Government work and not under copyright protection in the US; foreign copyright protection may apply.
Conflict of interest statement
The authors declare no competing interests.
Figures
Update of
-
QTL mapping of human retina DNA methylation identifies 87 gene-epigenome interactions in age-related macular degeneration.Res Sq [Preprint]. 2023 Jun 16:rs.3.rs-3011096. doi: 10.21203/rs.3.rs-3011096/v1. Res Sq. 2023. Update in: Nat Commun. 2024 Mar 4;15(1):1972. doi: 10.1038/s41467-024-46063-8. PMID: 37398472 Free PMC article. Updated. Preprint.
References
-
- Frydas, A., Wauters, E., van der Zee, J. & Van Broeckhoven, C. Uncovering the impact of noncoding variants in neurodegenerative brain diseases. Trends Genet.38, 258–272 (2022). - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
