

Population-level comparisons of gene regulatory networks modeled on high-throughput single-cell transcriptomics data
- PMID: 38438786
- PMCID: PMC10965443
- DOI: 10.1038/s43588-024-00597-5
Population-level comparisons of gene regulatory networks modeled on high-throughput single-cell transcriptomics data
Abstract
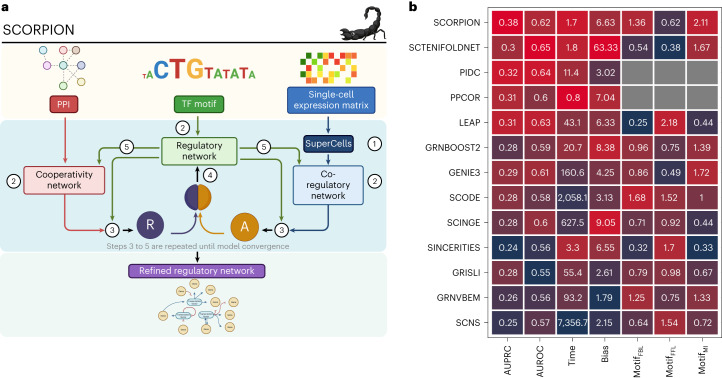
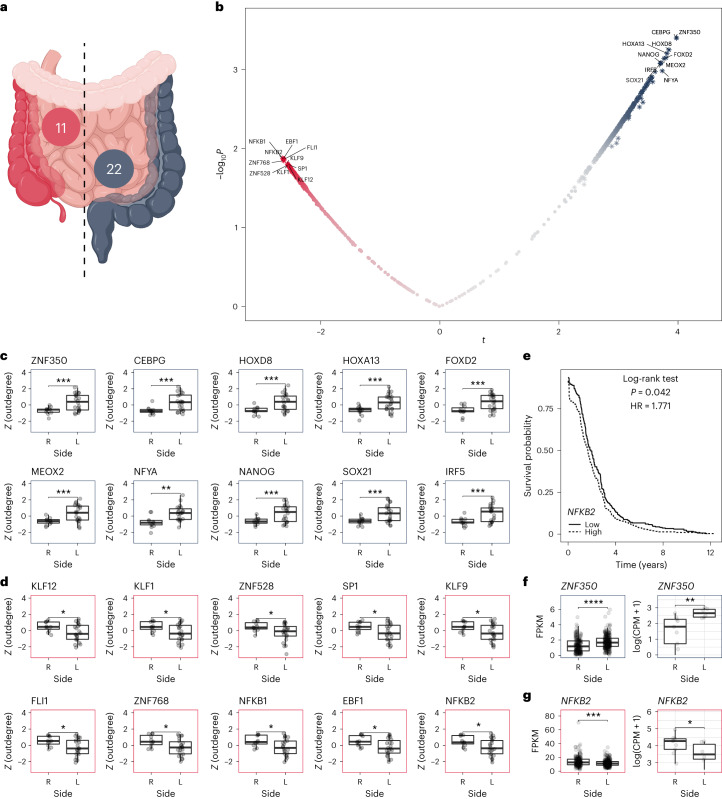
Single-cell technologies enable high-resolution studies of phenotype-defining molecular mechanisms. However, data sparsity and cellular heterogeneity make modeling biological variability across single-cell samples difficult. Here we present SCORPION, a tool that uses a message-passing algorithm to reconstruct comparable gene regulatory networks from single-cell/nuclei RNA-sequencing data that are suitable for population-level comparisons by leveraging the same baseline priors. Using synthetic data, we found that SCORPION outperformed 12 existing gene regulatory network reconstruction techniques. Using supervised experiments, we show that SCORPION can accurately identify differences in regulatory networks between wild-type and transcription factor-perturbed cells. We demonstrate SCORPION's scalability to population-level analyses using a single-cell RNA-sequencing atlas containing 200,436 cells from colorectal cancer and adjacent healthy tissues. The differences between tumor regions detected by SCORPION are consistent across multiple cohorts as well as with our understanding of disease progression, and elucidate phenotypic regulators that may impact patient survival.
© 2024. The Author(s).
Conflict of interest statement
D.O. is currently an employee of QIAGEN Digital Insights, QIAGEN, USA. The other authors declare no competing interests.
Figures




References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
