

Expanded clinical phenotype and untargeted metabolomics analysis in RARS2-related mitochondrial disorder: a case report
- PMID: 38438854
- PMCID: PMC10910770
- DOI: 10.1186/s12883-024-03571-w
Expanded clinical phenotype and untargeted metabolomics analysis in RARS2-related mitochondrial disorder: a case report
Abstract
Background: RARS2-related mitochondrial disorder is an autosomal recessive mitochondrial encephalopathy caused by biallelic pathogenic variants in the gene encoding the mitochondrial arginyl-transfer RNA synthetase 2 (RARS2, MIM *611524, NM_020320.5). RARS2 catalyzes the transfer of L-arginine to its cognate tRNA during the translation of mitochondrially-encoded proteins. The classical presentation of RARS2-related mitochondrial disorder includes pontocerebellar hypoplasia (PCH), progressive microcephaly, profound developmental delay, feeding difficulties, and hypotonia. Most patients also develop severe epilepsy by three months of age, which consists of focal or generalized seizures that frequently become pharmacoresistant and lead to developmental and epileptic encephalopathy (DEE).
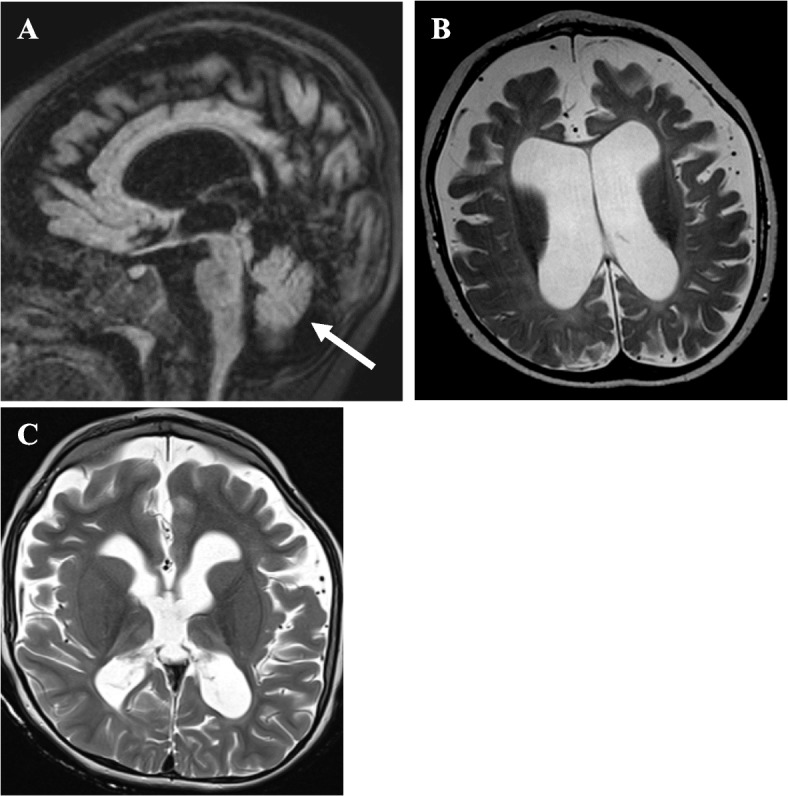
Case presentation: Here, we describe a six-year-old boy with developmental delay, hypotonia, and failure to thrive who developed an early-onset DEE consistent with Lennox-Gastaut Syndrome (LGS), which has not previously been observed in this disorder. He had dysmorphic features including bilateral macrotia, overriding second toes, a depressed nasal bridge, retrognathia, and downslanting palpebral fissures, and he did not demonstrate progressive microcephaly. Whole genome sequencing identified two variants in RARS2, c.36 + 1G > T, a previously unpublished variant that is predicted to affect splicing and is, therefore, likely pathogenic and c.419 T > G (p.Phe140Cys), a known pathogenic variant. He exhibited significant, progressive generalized brain atrophy and ex vacuo dilation of the supratentorial ventricular system on brain MRI and did not demonstrate PCH. Treatment with a ketogenic diet (KD) reduced seizure frequency and enabled him to make developmental progress. Plasma untargeted metabolomics analysis showed increased levels of lysophospholipid and sphingomyelin-related metabolites.
Conclusions: Our work expands the clinical spectrum of RARS2-related mitochondrial disorder, demonstrating that patients can present with dysmorphic features and an absence of progressive microcephaly, which can help guide the diagnosis of this condition. Our case highlights the importance of appropriate seizure phenotyping in this condition and indicates that patients can develop LGS, for which a KD may be a viable therapeutic option. Our work further suggests that analytes of phospholipid metabolism may serve as biomarkers of mitochondrial dysfunction.
Keywords: RARS2; Dysmorphic features; Lennox-Gastaut Syndrome; Mitochondrial disease; Untargeted metabolomics analysis.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Joseph JT, Innes AM, Smith AC, Vanstone MR, Schwartzentruber JA, Bulman DE, Majewski J, Daza RA, Hevner RF, Michaud J, Boycott KM; FORGE Canada Consortium. Neuropathologic features of pontocerebellar hypoplasia type 6. J Neuropathol Exp Neurol. 2014;73(11):1009–25. 10.1097/NEN.0000000000000123. - PubMed
-
- Glamuzina E, Brown R, Hogarth K, Saunders D, Russell-Eggitt I, Pitt M, de Sousa C, Rahman S, Brown G, Grunewald S. Further delineation of pontocerebellar hypoplasia type 6 due to mutations in the gene encoding mitochondrial arginyl-tRNA synthetase, RARS2. J Inherit Metab Dis. 2012;35(3):459–467. doi: 10.1007/s10545-011-9413-6. - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
