

Emergence and spread of Mycobacterium ulcerans at different geographic scales
- PMID: 38441471
- PMCID: PMC10986537
- DOI: 10.1128/spectrum.03827-23
Emergence and spread of Mycobacterium ulcerans at different geographic scales
Abstract
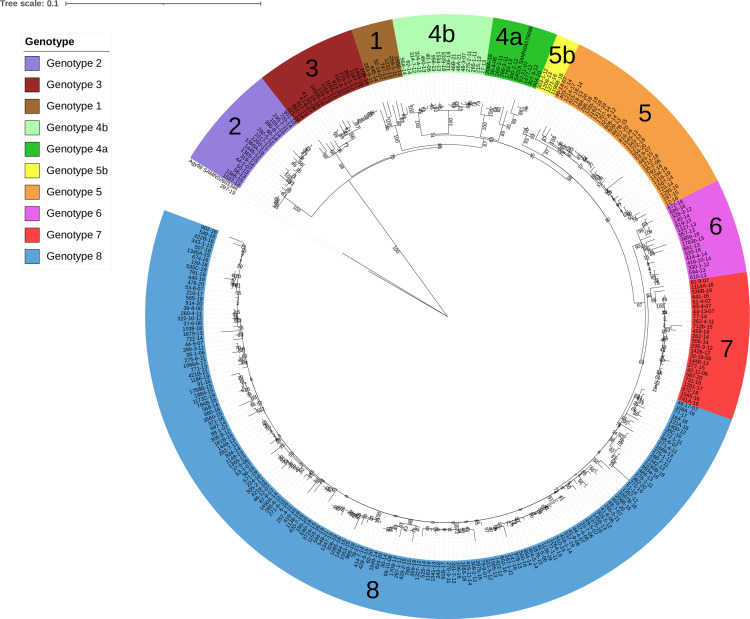
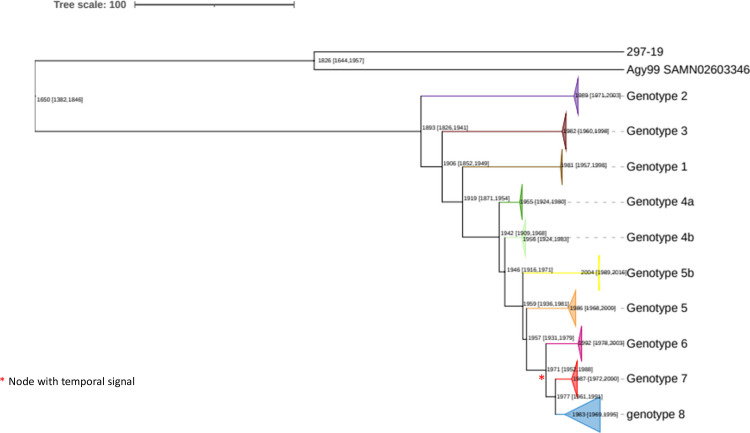
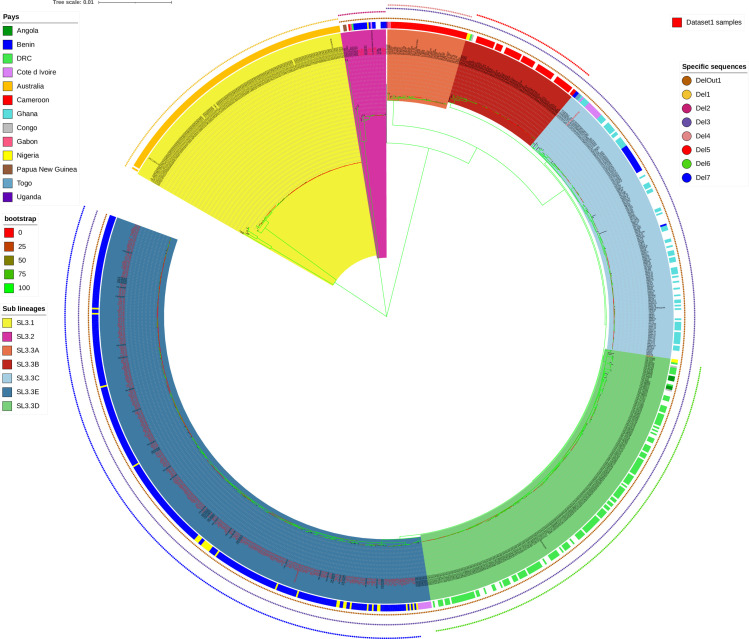
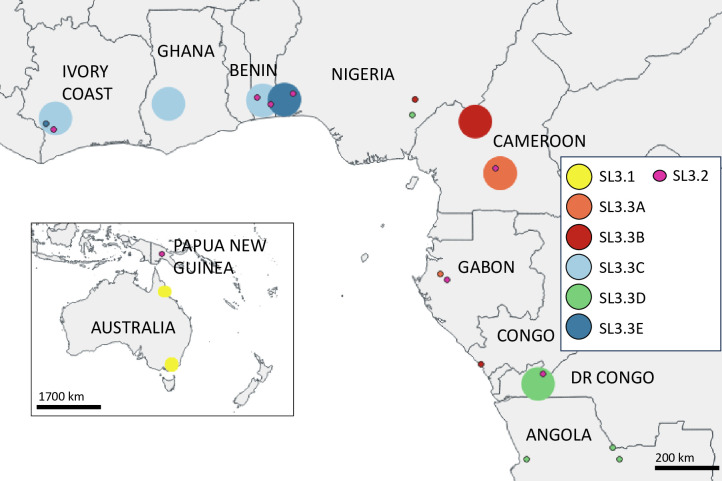
The classical lineage of Mycobacterium ulcerans is the most prevalent clonal group associated with Buruli ulcer in humans. Its reservoir is strongly associated with the environment. We analyzed together 1,045 isolates collected from 13 countries on two continents to define the evolutionary history and population dynamics of this lineage. We confirm that this lineage spread over 7,000 years from Australia to Africa with the emergence of outbreaks in distinct waves in the 18th and 19th centuries. In sharp contrast with its global spread over the last century, transmission chains are now mostly local, with little or no dissemination between endemic areas. This study provides new insights into the phylogeography and population dynamics of M. ulcerans, highlighting the importance of comparative genomic analyses to improve our understanding of pathogen transmission.
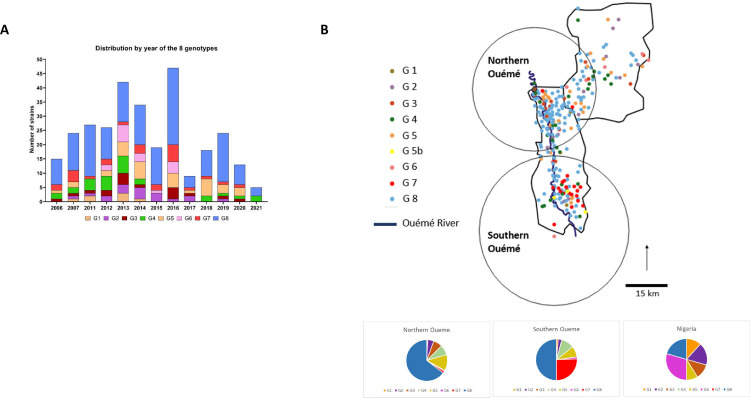
Importance: Mycobacterium ulcerans is an environmental mycobacterial pathogen that can cause Buruli ulcer, a severe cutaneous infection, mostly spread in Africa and Australia. We conducted a large genomic study of M. ulcerans, combining genomic and evolutionary approaches to decipher its evolutionary history and pattern of spread at different geographic scales. At the scale of villages in an endemic area of Benin, the circulating genotypes have been introduced in recent decades and are not randomly distributed along the river. On a global scale, M. ulcerans has been spreading for much longer, resulting in distinct and compartmentalized endemic foci across Africa and Australia.
Keywords: Mycobacterium ulcerans; WGS; evolution; phylogeography.
Conflict of interest statement
The authors declare no conflict of interest.
Figures



Similar articles
-
Mycobacterium ulcerans Population Genomics To Inform on the Spread of Buruli Ulcer across Central Africa.mSphere. 2019 Feb 6;4(1):e00472-18. doi: 10.1128/mSphere.00472-18. mSphere. 2019. PMID: 30728280 Free PMC article.
-
Comparative Genomics Shows That Mycobacterium ulcerans Migration and Expansion Preceded the Rise of Buruli Ulcer in Southeastern Australia.Appl Environ Microbiol. 2018 Apr 2;84(8):e02612-17. doi: 10.1128/AEM.02612-17. Print 2018 Apr 15. Appl Environ Microbiol. 2018. PMID: 29439984 Free PMC article.
-
Stable and Local Reservoirs of Mycobacterium ulcerans Inferred from the Nonrandom Distribution of Bacterial Genotypes, Benin.Emerg Infect Dis. 2020 Mar;26(3):491-503. doi: 10.3201/eid2603.190573. Emerg Infect Dis. 2020. PMID: 32091371 Free PMC article.
-
Buruli ulcer: reductive evolution enhances pathogenicity of Mycobacterium ulcerans.Nat Rev Microbiol. 2009 Jan;7(1):50-60. doi: 10.1038/nrmicro2077. Nat Rev Microbiol. 2009. PMID: 19079352 Review.
-
Understanding the transmission of Mycobacterium ulcerans: A step towards controlling Buruli ulcer.PLoS Negl Trop Dis. 2021 Aug 26;15(8):e0009678. doi: 10.1371/journal.pntd.0009678. eCollection 2021 Aug. PLoS Negl Trop Dis. 2021. PMID: 34437549 Free PMC article. Review.
Cited by
-
Analysis of Agro-Environmental and Geo-Climatic Factors Influencing Buruli Ulcer in the Kimpese Health Zone in Kongo Central, Democratic Republic of Congo.Public Health Chall. 2025 Aug 12;4(3):e70111. doi: 10.1002/puh2.70111. eCollection 2025 Sep. Public Health Chall. 2025. PMID: 40800030 Free PMC article.
-
Spatiotemporal distribution of Mycobacterium ulcerans and other mycolactone producing mycobacteria in southeastern United States.Emerg Microbes Infect. 2025 Dec;14(1):2521853. doi: 10.1080/22221751.2025.2521853. Epub 2025 Jul 2. Emerg Microbes Infect. 2025. PMID: 40525224 Free PMC article.
References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
