

Integrating high-throughput analysis to create an atlas of replication origins in Trypanosoma cruzi in the context of genome structure and variability
- PMID: 38441981
- PMCID: PMC11005370
- DOI: 10.1128/mbio.00319-24
Integrating high-throughput analysis to create an atlas of replication origins in Trypanosoma cruzi in the context of genome structure and variability
Abstract
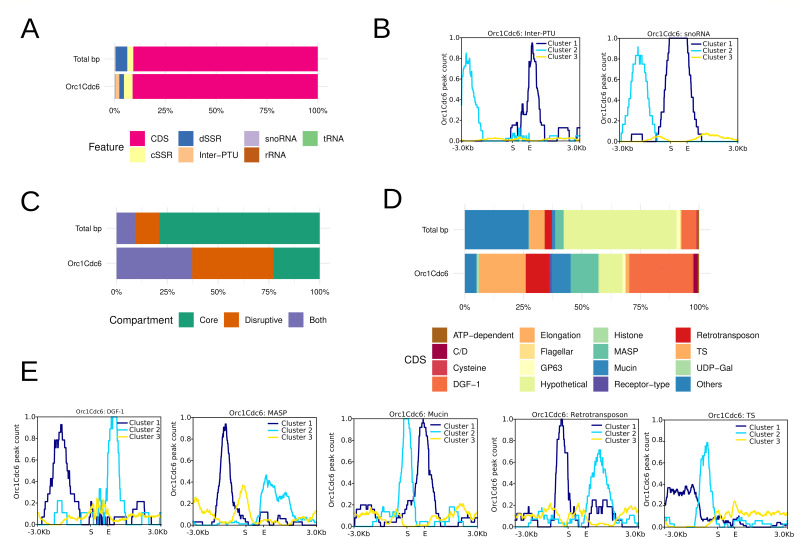
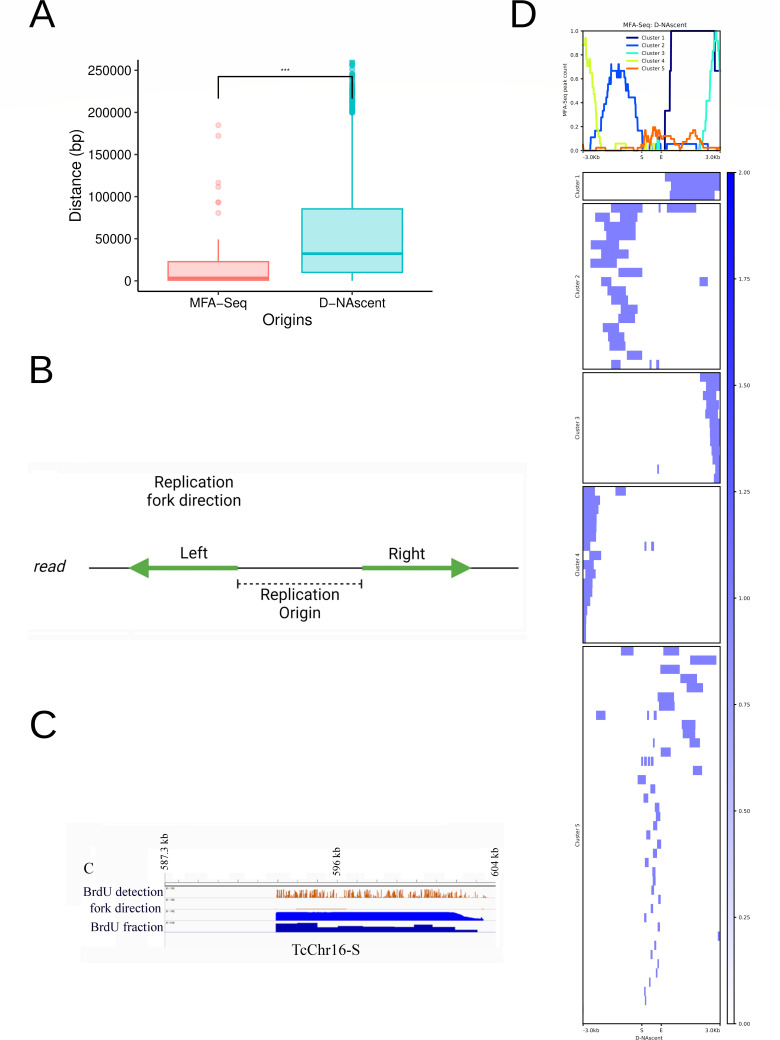
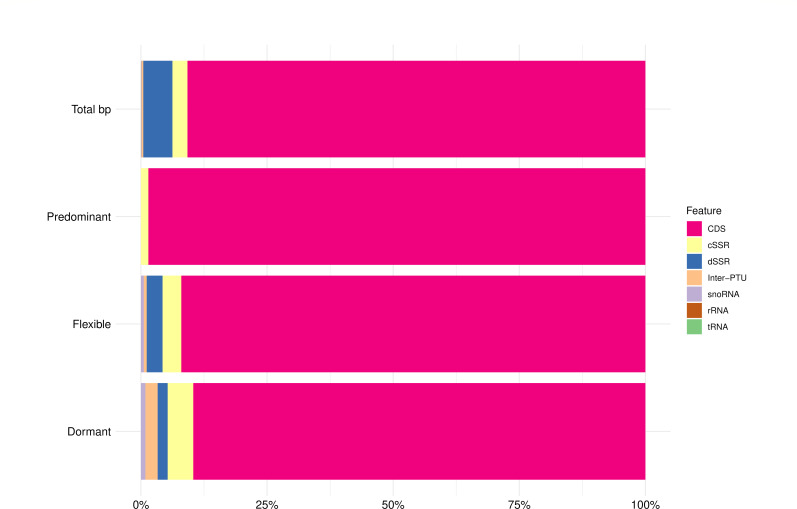
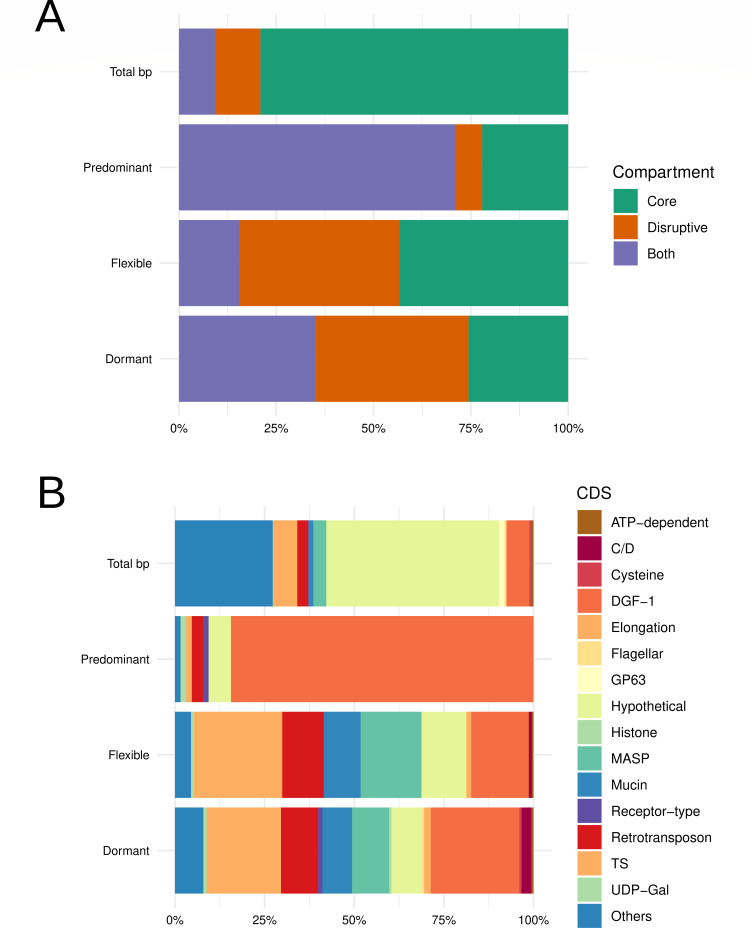
Trypanosoma cruzi is the etiologic agent of the most prevalent human parasitic disease in Latin America, Chagas disease. Its genome is rich in multigenic families that code for virulent antigens and are present in the rapidly evolving genomic compartment named Disruptive. DNA replication is a meticulous biological process in which flaws can generate mutations and changes in chromosomal and gene copy numbers. Here, integrating high-throughput and single-molecule analyses, we were able to identify Predominant, Flexible, and Dormant Orc1Cdc6-dependent origins as well as Orc1Cdc6-independent origins. Orc1Cdc6-dependent origins were found in multigenic family loci, while independent origins were found in the Core compartment that contains conserved and hypothetical protein-coding genes, in addition to multigenic families. In addition, we found that Orc1Cdc6 density is related to the firing of origins and that Orc1Cdc6-binding sites within fired origins are depleted of a specific class of nucleosomes that we previously categorized as dynamic. Together, these data suggest that Orc1Cdc6-dependent origins may contribute to the rapid evolution of the Disruptive compartment and, therefore, to the success of T. cruzi infection and that the local epigenome landscape is also involved in this process.IMPORTANCETrypanosoma cruzi, responsible for Chagas disease, affects millions globally, particularly in Latin America. Lack of vaccine or treatment underscores the need for research. Parasite's genome, with virulent antigen-coding multigenic families, resides in the rapidly evolving Disruptive compartment. Study sheds light on the parasite's dynamic DNA replication, discussing the evolution of the Disruptive compartment. Therefore, the findings represent a significant stride in comprehending T. cruzi's biology and the molecular bases that contribute to the success of infection caused by this parasite.
Keywords: DNA replication; Trypanosoma cruzi; prereplication complex; replication origins.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Expanding an expanded genome: long-read sequencing of Trypanosoma cruzi.Microb Genom. 2018 May;4(5):e000177. doi: 10.1099/mgen.0.000177. Epub 2018 Apr 30. Microb Genom. 2018. PMID: 29708484 Free PMC article.
-
Accessing the Variability of Multicopy Genes in Complex Genomes using Unassembled Next-Generation Sequencing Reads: The Case of Trypanosoma cruzi Multigene Families.mBio. 2022 Dec 20;13(6):e0231922. doi: 10.1128/mbio.02319-22. Epub 2022 Oct 20. mBio. 2022. PMID: 36264102 Free PMC article.
-
Replication origin location might contribute to genetic variability in Trypanosoma cruzi.BMC Genomics. 2020 Jun 22;21(1):414. doi: 10.1186/s12864-020-06803-8. BMC Genomics. 2020. PMID: 32571205 Free PMC article.
-
Trypanosoma cruzi: Genomic Diversity and Structure.Pathogens. 2025 Jan 12;14(1):61. doi: 10.3390/pathogens14010061. Pathogens. 2025. PMID: 39861022 Free PMC article. Review.
-
Trypanosoma Cruzi Genome: Organization, Multi-Gene Families, Transcription, and Biological Implications.Genes (Basel). 2020 Oct 14;11(10):1196. doi: 10.3390/genes11101196. Genes (Basel). 2020. PMID: 33066599 Free PMC article. Review.
References
-
- Zingales B, Andrade SG, Briones MRS, Campbell DA, Chiari E, Fernandes O, Guhl F, Lages-Silva E, Macedo AM, Machado CR, Miles MA, Romanha AJ, Sturm NR, Tibayrenc M, Schijman AG, Second Satellite Meeting . 2009. A new consensus for Trypanosoma cruzi intraspecific nomenclature: second revision meeting recommends TcI to TcVI. Mem Inst Oswaldo Cruz 104:1051–1054. doi:10.1590/s0074-02762009000700021 - DOI - PubMed
-
- Reis-Cunha JL, Baptista RP, Rodrigues-Luiz GF, Coqueiro-Dos-Santos A, Valdivia HO, de Almeida LV, Cardoso MS, D’Ávila DA, Dias FHC, Fujiwara RT, Galvão LMC, Chiari E, Cerqueira GC, Bartholomeu DC. 2018. Whole genome sequencing of Trypanosoma cruzi field isolates reveals extensive genomic variability and complex aneuploidy patterns within TcII DTU. BMC Genomics 19:816. doi:10.1186/s12864-018-5198-4 - DOI - PMC - PubMed
MeSH terms
Grants and funding
- 2013/07467-1/Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP)
- 2016/50050-2/Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP)
- 2017/07693-2/Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP)
- 2019/04483-2/Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP)
- 2020/00694-6/Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP)
LinkOut - more resources
Full Text Sources
Medical
