

NADPH oxidase exerts a B cell-intrinsic contribution to lupus risk by modulating endosomal TLR signals
- PMID: 38442270
- PMCID: PMC10913815
- DOI: 10.1084/jem.20230774
NADPH oxidase exerts a B cell-intrinsic contribution to lupus risk by modulating endosomal TLR signals
Abstract
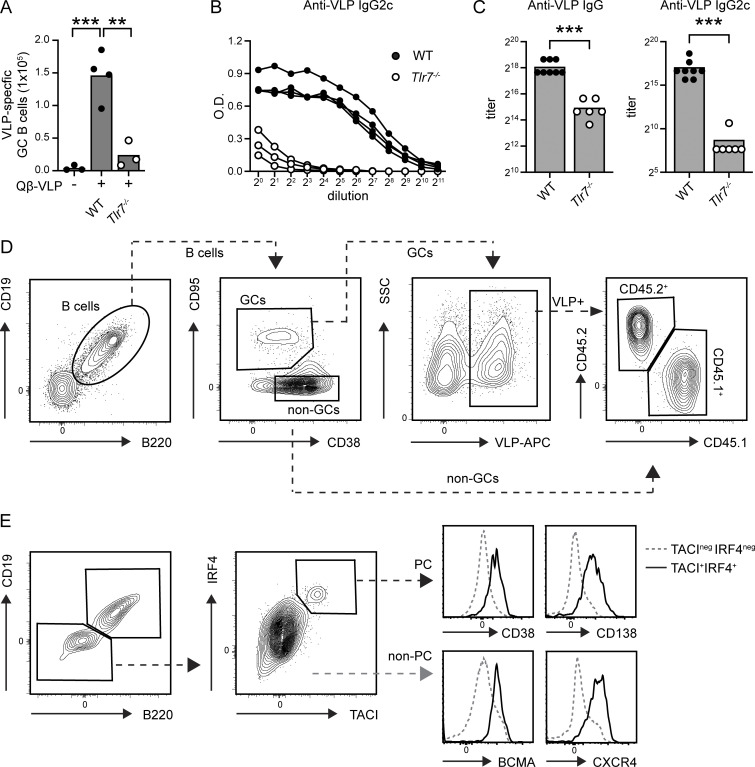
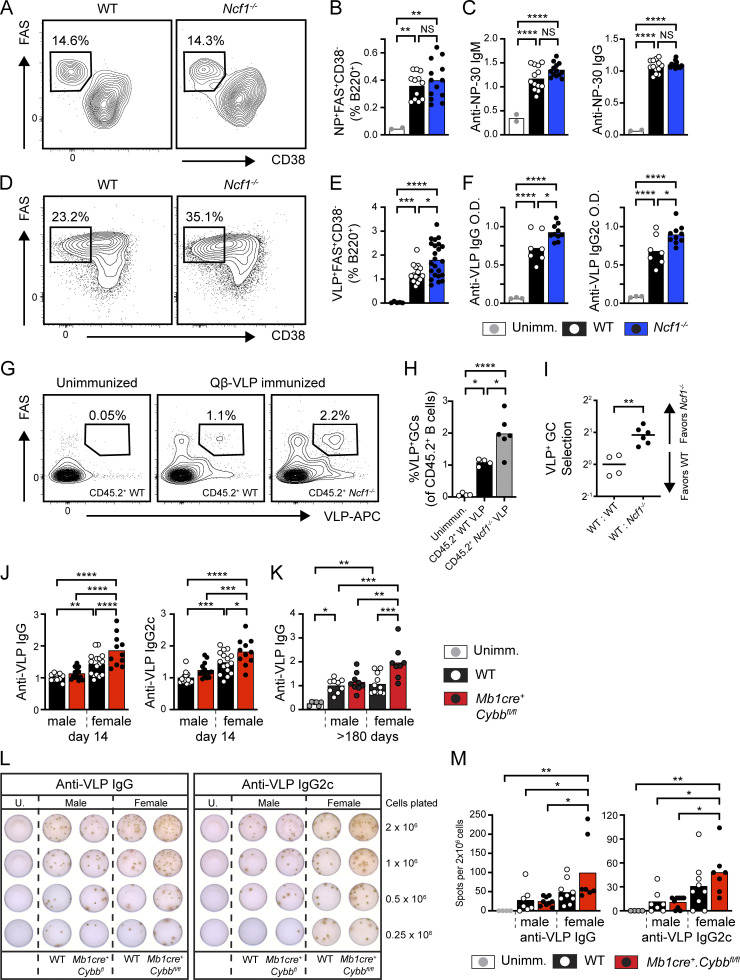
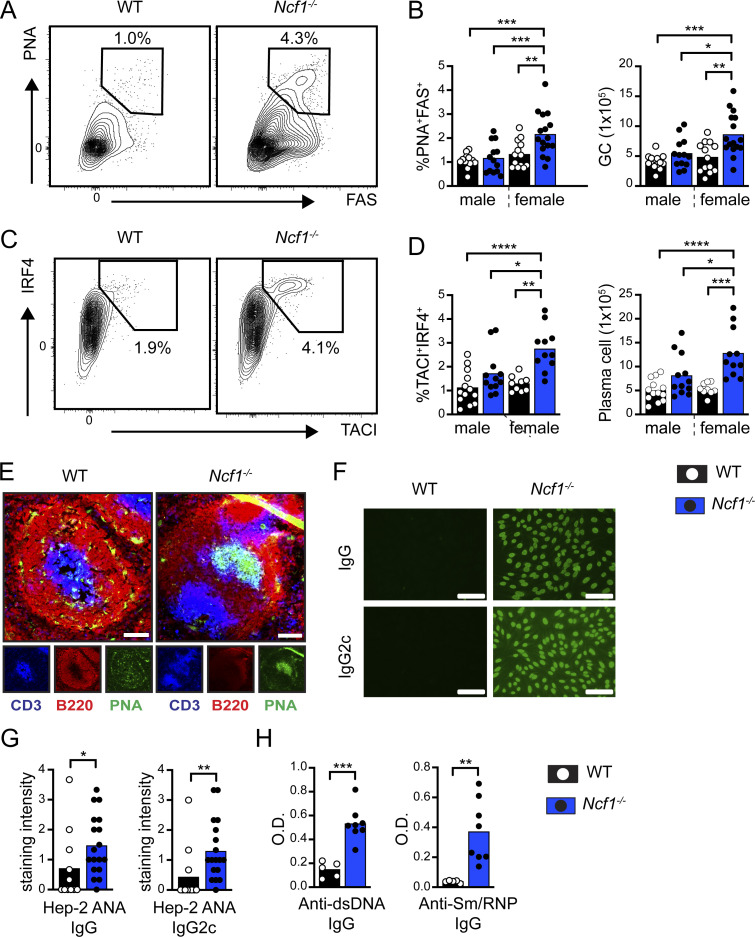
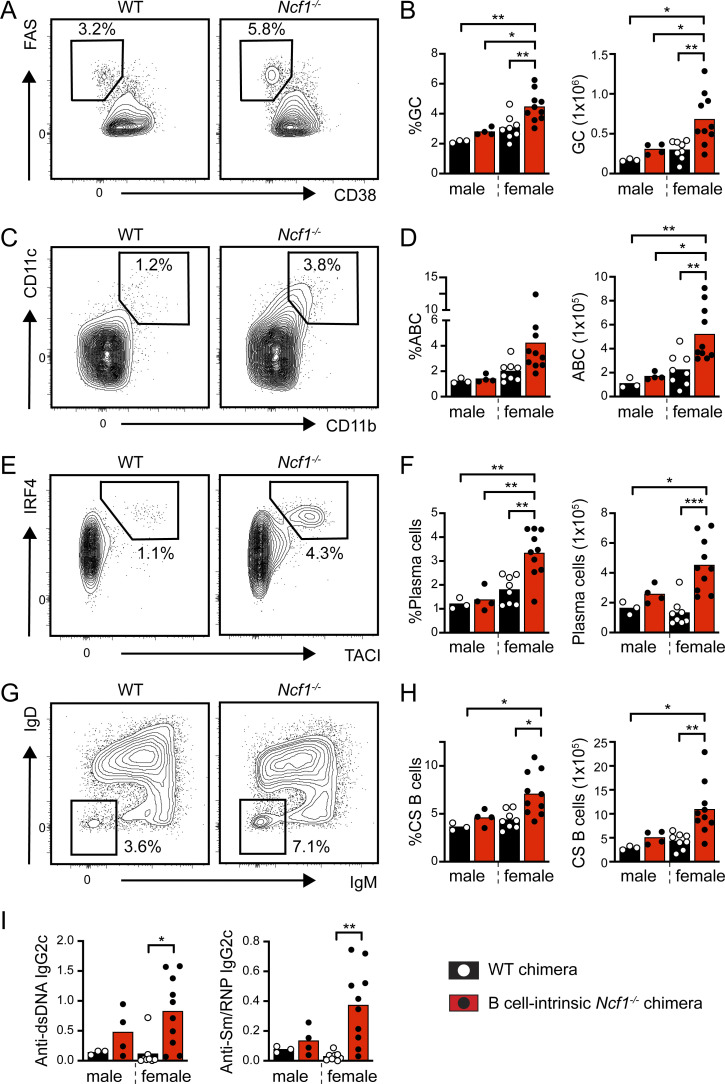
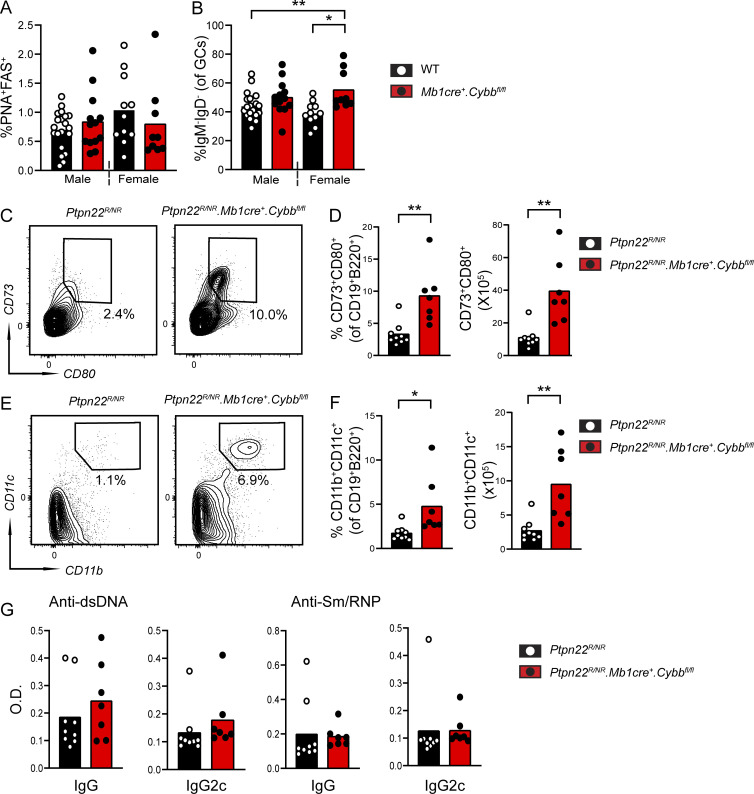
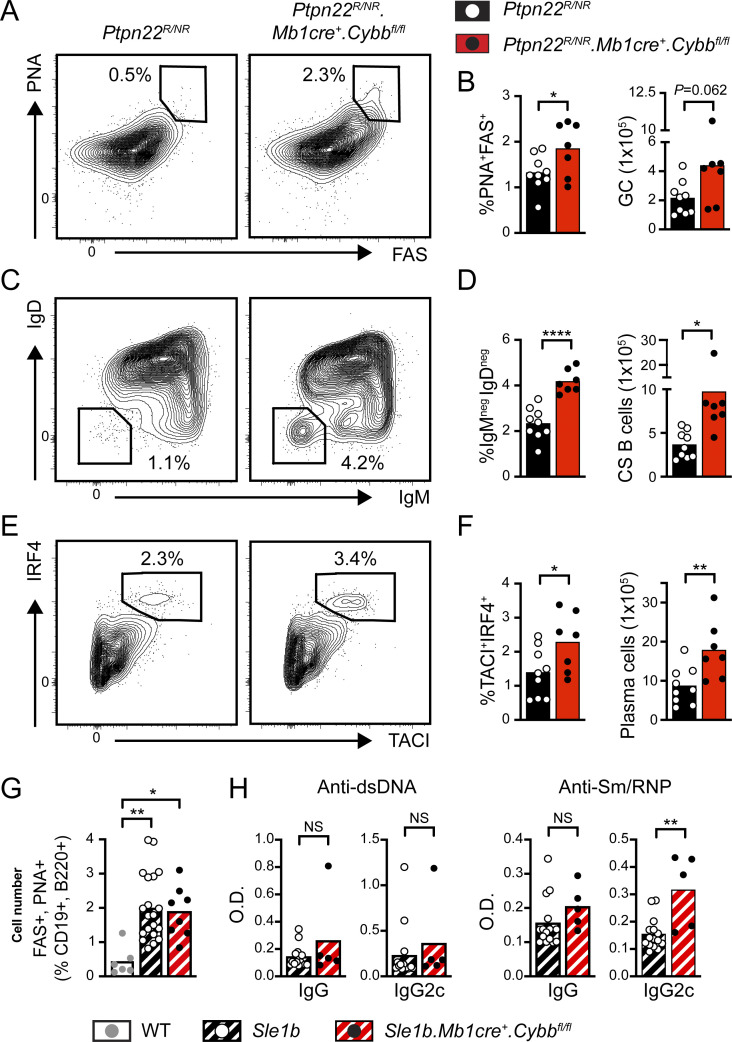
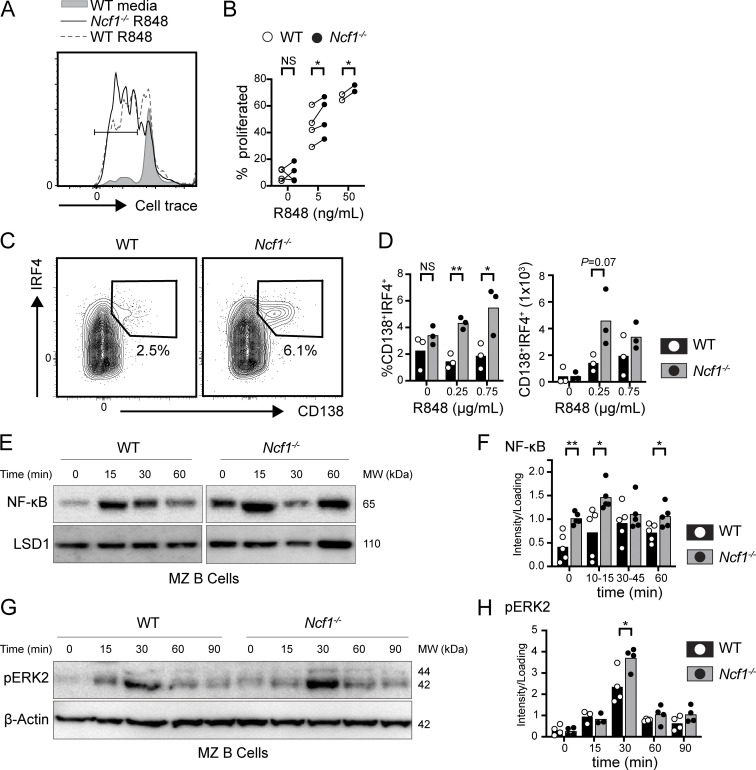
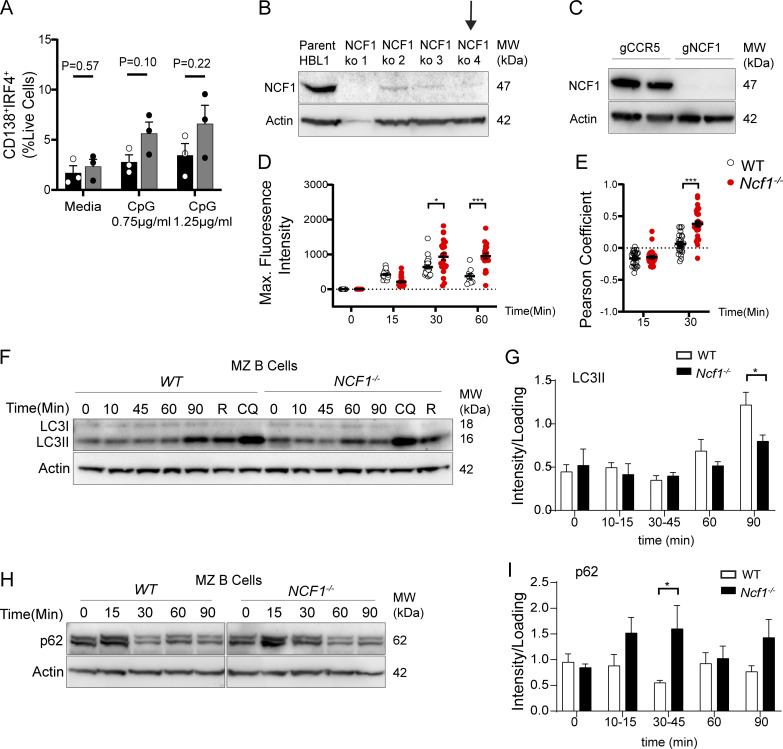
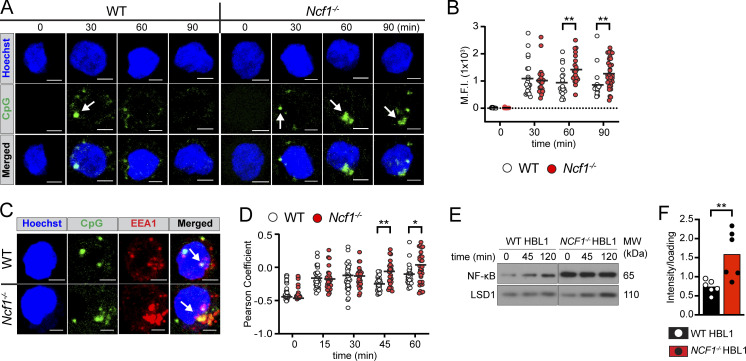
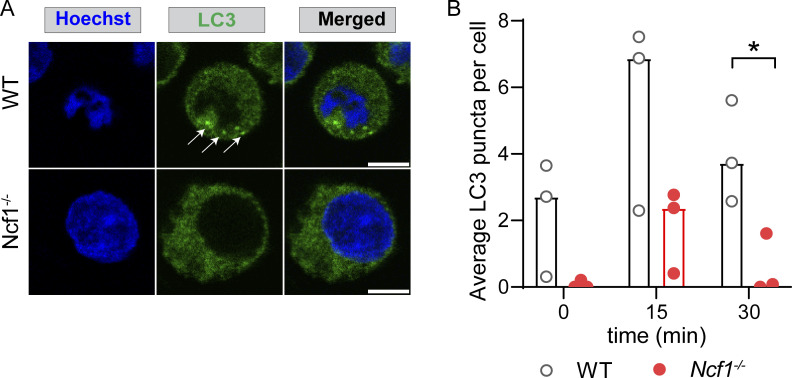
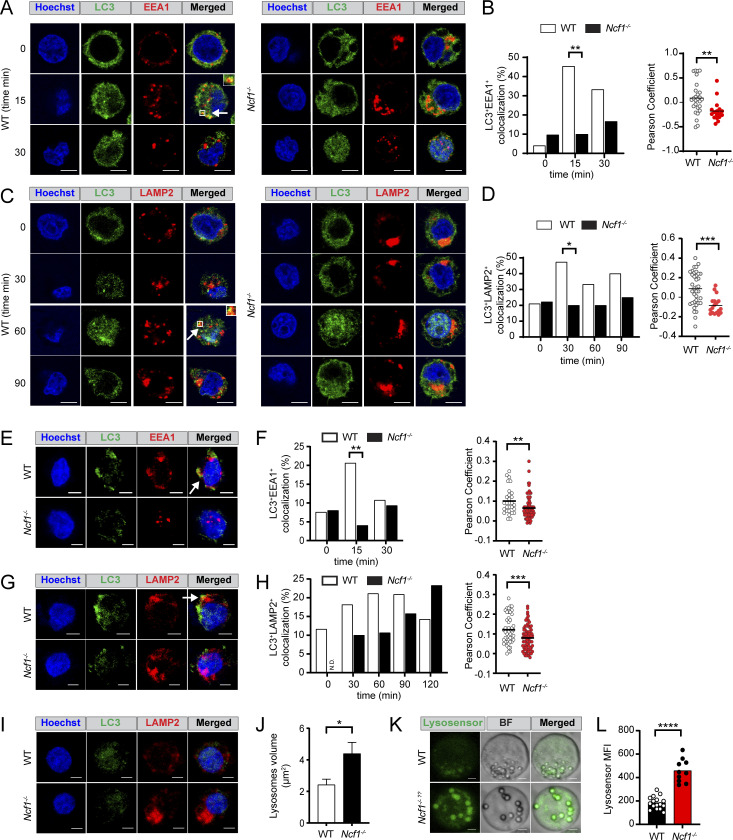
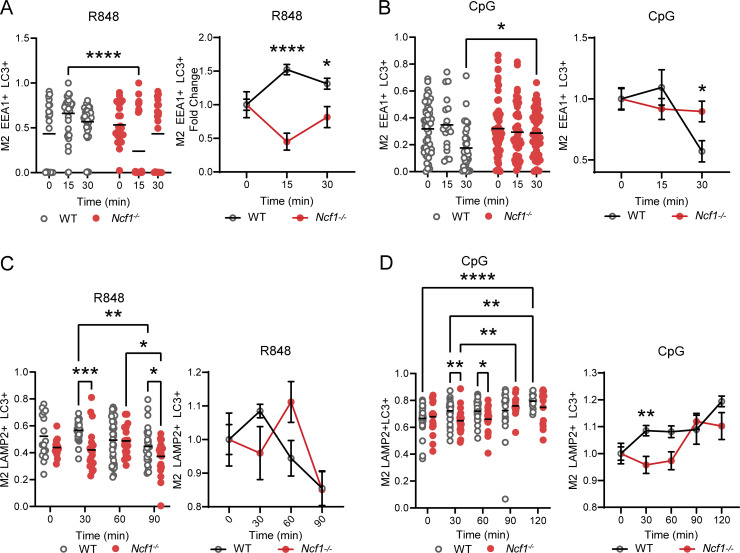
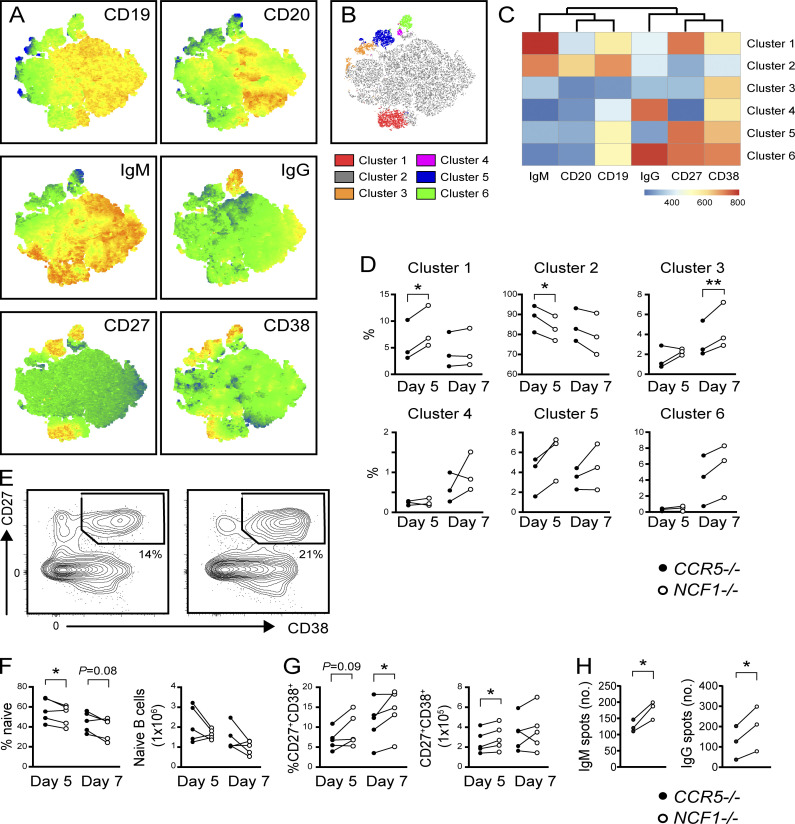
Genome-wide association studies in systemic lupus erythematosus (SLE) have linked loss-of-function mutations in phagocytic NADPH oxidase complex (NOX2) genes, including NCF1 and NCF2, to disease pathogenesis. The prevailing model holds that reduced NOX2 activity promotes SLE via defective efferocytosis, the immunologically silent clearance of apoptotic cells. Here, we describe a parallel B cell-intrinsic mechanism contributing to breaks in tolerance. In keeping with an important role for B cell Toll-like receptor (TLR) pathways in lupus pathogenesis, NOX2-deficient B cells exhibit enhanced signaling downstream of endosomal TLRs, increased humoral responses to nucleic acid-containing antigens, and the propensity toward humoral autoimmunity. Mechanistically, TLR-dependent NOX2 activation promotes LC3-mediated maturation of TLR-containing endosomes, resulting in signal termination. CRISPR-mediated disruption of NCF1 confirmed a direct role for NOX2 in regulating endosomal TLR signaling in primary human B cells. Together, these data highlight a new B cell-specific mechanism contributing to autoimmune risk in NCF1 and NCF2 variant carriers.
© 2024 Liu et al.
Conflict of interest statement
Disclosures: S.W. Jackson reported personal fees from Bristol-Myers Squib during the conduct of the study; and personal fees from ChemoCentryx, Inc. and Variant Bio outside the submitted work. No other disclosures were reported.
Figures
References
-
- Alarcón-Riquelme, M.E., Ziegler J.T., Molineros J., Howard T.D., Moreno-Estrada A., Sánchez-Rodríguez E., Ainsworth H.C., Ortiz-Tello P., Comeau M.E., Rasmussen A., et al. . 2016. Genome-wide association study in an amerindian ancestry population reveals novel systemic lupus erythematosus risk loci and the role of European admixture. Arthritis Rheumatol. 68:932–943. 10.1002/art.39504 - DOI - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
