

SPREd: a simulation-supervised neural network tool for gene regulatory network reconstruction
- PMID: 38444538
- PMCID: PMC10913396
- DOI: 10.1093/bioadv/vbae011
SPREd: a simulation-supervised neural network tool for gene regulatory network reconstruction
Abstract
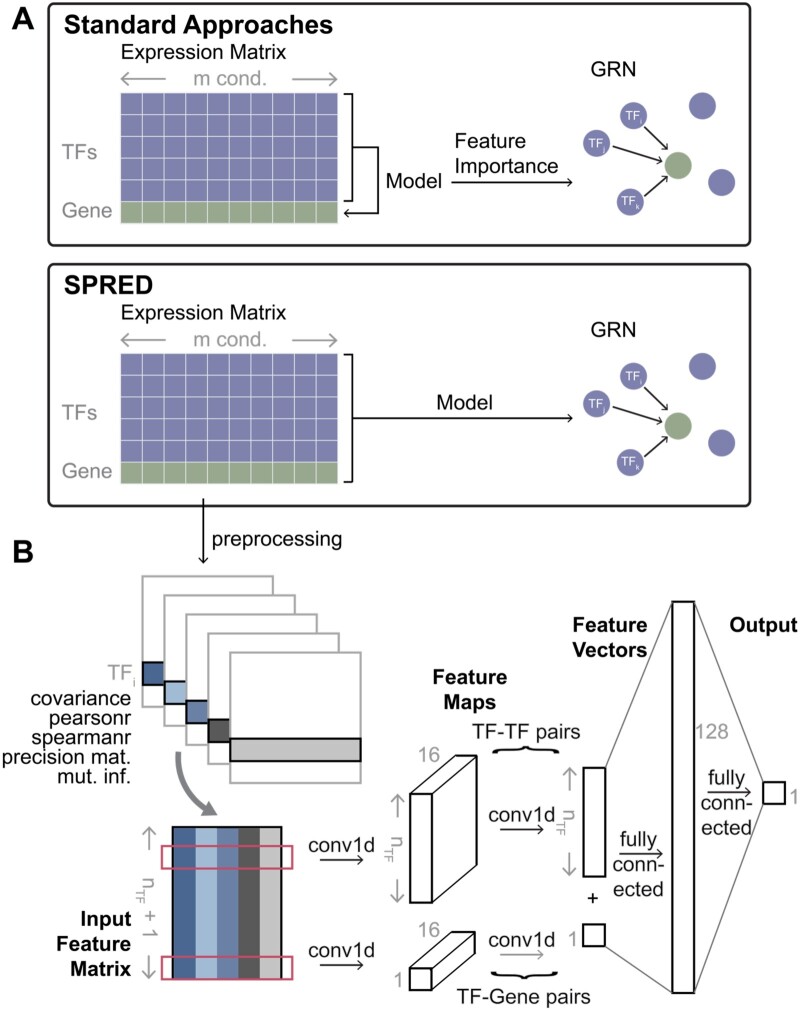
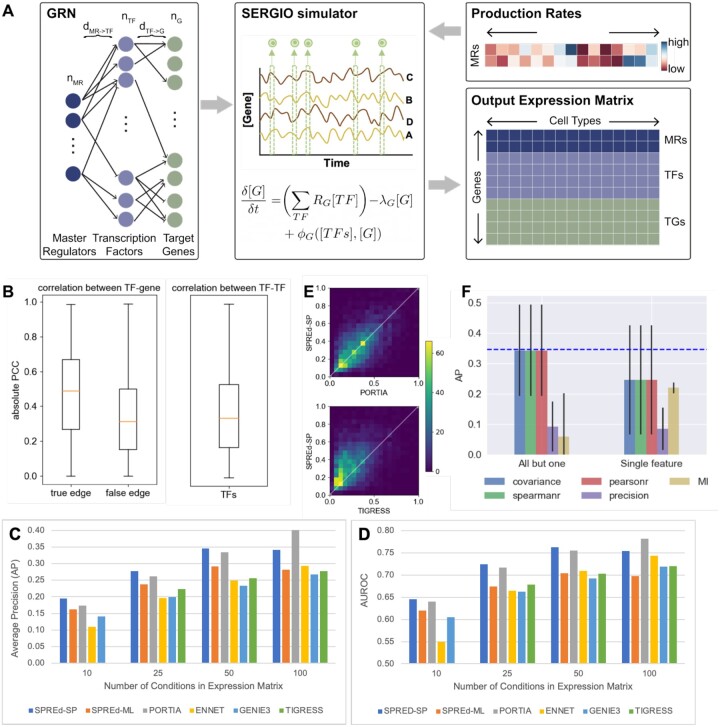
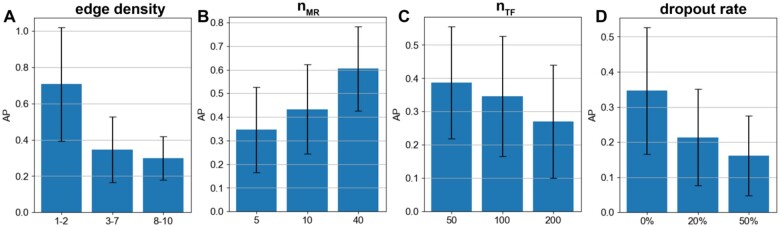
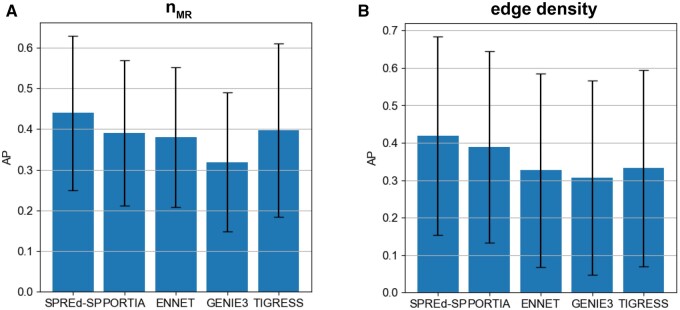
Summary: Reconstruction of gene regulatory networks (GRNs) from expression data is a significant open problem. Common approaches train a machine learning (ML) model to predict a gene's expression using transcription factors' (TFs') expression as features and designate important features/TFs as regulators of the gene. Here, we present an entirely different paradigm, where GRN edges are directly predicted by the ML model. The new approach, named "SPREd," is a simulation-supervised neural network for GRN inference. Its inputs comprise expression relationships (e.g. correlation, mutual information) between the target gene and each TF and between pairs of TFs. The output includes binary labels indicating whether each TF regulates the target gene. We train the neural network model using synthetic expression data generated by a biophysics-inspired simulation model that incorporates linear as well as non-linear TF-gene relationships and diverse GRN configurations. We show SPREd to outperform state-of-the-art GRN reconstruction tools GENIE3, ENNET, PORTIA, and TIGRESS on synthetic datasets with high co-expression among TFs, similar to that seen in real data. A key advantage of the new approach is its robustness to relatively small numbers of conditions (columns) in the expression matrix, which is a common problem faced by existing methods. Finally, we evaluate SPREd on real data sets in yeast that represent gold-standard benchmarks of GRN reconstruction and show it to perform significantly better than or comparably to existing methods. In addition to its high accuracy and speed, SPREd marks a first step toward incorporating biophysics principles of gene regulation into ML-based approaches to GRN reconstruction.
Availability and implementation: Data and code are available from https://github.com/iiiime/SPREd.
© The Author(s) 2024. Published by Oxford University Press.
Conflict of interest statement
None declared.
Figures
Update of
-
SPREd: A simulation-supervised neural network tool for gene regulatory network reconstruction.bioRxiv [Preprint]. 2023 Nov 13:2023.11.09.566399. doi: 10.1101/2023.11.09.566399. bioRxiv. 2023. Update in: Bioinform Adv. 2024 Jan 23;4(1):vbae011. doi: 10.1093/bioadv/vbae011. PMID: 38014297 Free PMC article. Updated. Preprint.
References
-
- Box GE, Cox DR.. An analysis of transformations. J R Stat Soc Ser B Stat Methodol 1964;26:211–43.
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
