

Hereditary angioedema with normal C1 inhibitor associated with carboxypeptidase N deficiency
- PMID: 38445235
- PMCID: PMC10912455
- DOI: 10.1016/j.jacig.2024.100223
Hereditary angioedema with normal C1 inhibitor associated with carboxypeptidase N deficiency
Erratum in
-
Corrigendum.J Allergy Clin Immunol Glob. 2024 Aug 2;3(4):100319. doi: 10.1016/j.jacig.2024.100319. eCollection 2024 Nov. J Allergy Clin Immunol Glob. 2024. PMID: 39239323 Free PMC article.
Abstract
Background: Hereditary angioedema (HAE) is a potentially life-threatening disorder characterized by recurrent episodes of subcutaneous or submucosal swelling. HAE with normal C1 inhibitor (HAE-nC1-INH) is an underdiagnosed condition. Although the association with genetic variants has been identified for some families, the genetic causes in many patients with HAE-nC1-INH remain unknown. The role of genes associated with bradykinin catabolism is not fully understood.
Objective: We sought to investigate the biological parameters and the genes related to kallikrein-kinin system in families with a clinical phenotype of HAE-nC1-INH and presenting with a carboxypeptidase N (CPN) deficiency.
Methods: This study includes 4 families presenting with HAE-nC1-INH and CPN deficiency. Patients' clinical records were examined, biological parameters of kallikrein-kinin system were measured, and genetics was analyzed by next-generation sequencing and Sanger sequencing. Predictive algorithms (Human Splicing Finder, Sorting Intolerant From Tolerant, Polymorphism Phenotyping v2, MutationTaster, and ClinPred) were used to classify variants as affecting splicing, as benign to deleterious, or as disease-causing.
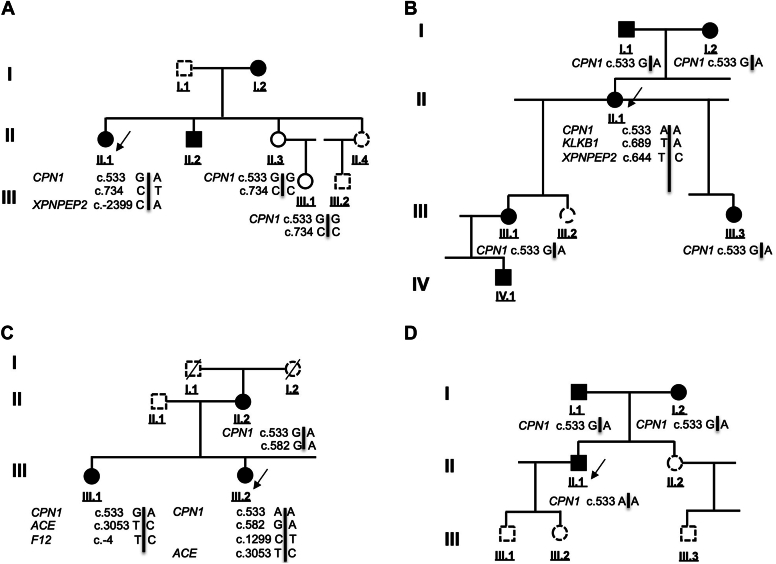
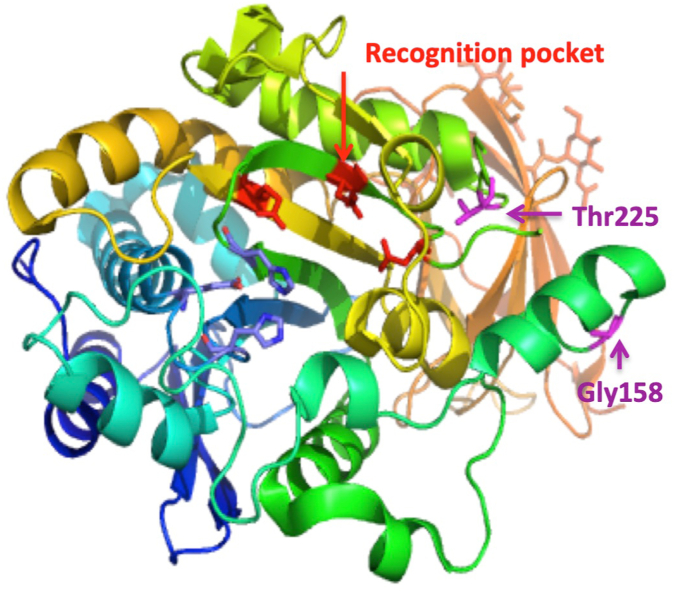
Results: Patients presented with angioedema and urticaria, mainly on face/lips, but also with abdominal pain or laryngeal symptoms. Affected patients displayed low CPN activity-30% to 50% of median value in plasma. We identified 3 variants of the CPN1 gene encoding the catalytic 55-kDa subunit of CPN: c.533G>A, c.582A>G, and c.734C>T. CPN deficiency associated with genetic variants segregated with HAE-nC1-INH symptoms in affected family members.
Conclusions: CPN1 gene variants are associated with CPN deficiency and HAE-nC1-INH symptoms in 4 unrelated families. Genetic CPN deficiency may contribute to bradykinin and anaphylatoxin accumulation, with synergistic effects in angioedema and urticarial symptoms.
Keywords: CPN1 gene; Urticaria; angioedema; hereditary carboxypeptidase N deficiency.
© 2024 The Author(s).
Conflict of interest statement
This work was supported by an 10.13039/100017732E-Rare-1 research grant attributed within European FP7 (HAEIII; S. Cichon, coordinator) and a French National Agency for Research grant (grant no. EudraCT #38RC09.023). The promoter for the study was CHU Grenoble Alpes (#2009-A00025-52). Funding was also obtained from the French National Blood Service (Etablissement Français du Sang) La Plaine Saint Denis (grant no. APR2016-64), from KininX SAS, and the National Rare Disease Program from the French Ministry of Health (National Reference Center for Angioedema CREAK). F.P. was recipient of a PhD fellowship from Etablissement Français du Sang (#APR2016-64). Disclosure of potential conflict of interest: F. Parsopoulou, G. Loules, and A. Ghannam received grants as stated in the funding section. The rest of the authors declare that they have no relevant conflicts of interest.
Figures
References
-
- Sharma J., Jindal A.K., Banday A.Z., Kaur A., Rawat A., Singh S., et al. Pathophysiology of hereditary angioedema (HAE) beyond the SERPING1 gene. Clin Rev Allergy Immunol. 2021;60:305–315. - PubMed
-
- Zanichelli A., Longhurst H.J., Maurer M., Bouillet L., Aberer W., Fabien V., et al. Misdiagnosis trends in patients with hereditary angioedema from the real-world clinical setting. Ann Allergy Asthma Immunol. 2016;117:394–398. - PubMed
-
- Dewald G., Bork K. Missense mutations in the coagulation factor XII (Hageman factor) gene in hereditary angioedema with normal C1 inhibitor. Biochem Biophys Res Commun. 2006;343:1286–1289. - PubMed
-
- Bork K., Wulff K., Steinmüller-Magin L., Braenne I., Staubach-Renz P., Witzke G., et al. Hereditary angioedema with a mutation in the plasminogen gene. Allergy. 2018;73:442–450. - PubMed
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
