

Strengthening Bordetella pertussis genomic surveillance by direct sequencing of residual positive specimens
- PMID: 38445858
- PMCID: PMC11005353
- DOI: 10.1128/jcm.01653-23
Strengthening Bordetella pertussis genomic surveillance by direct sequencing of residual positive specimens
Abstract
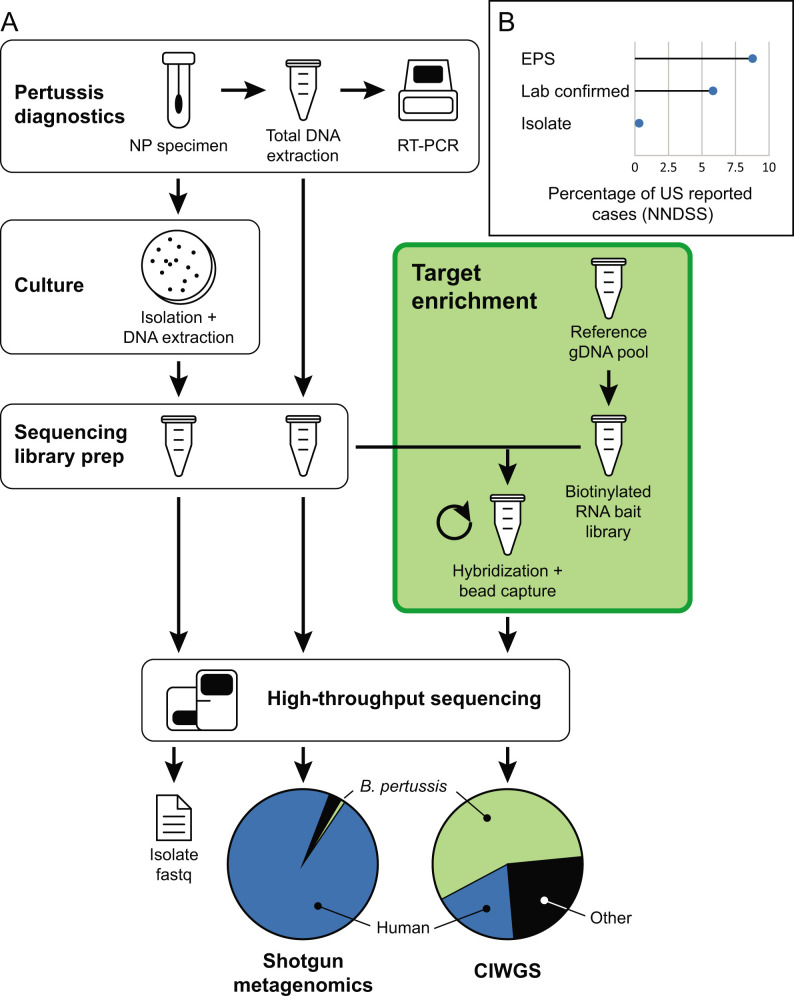
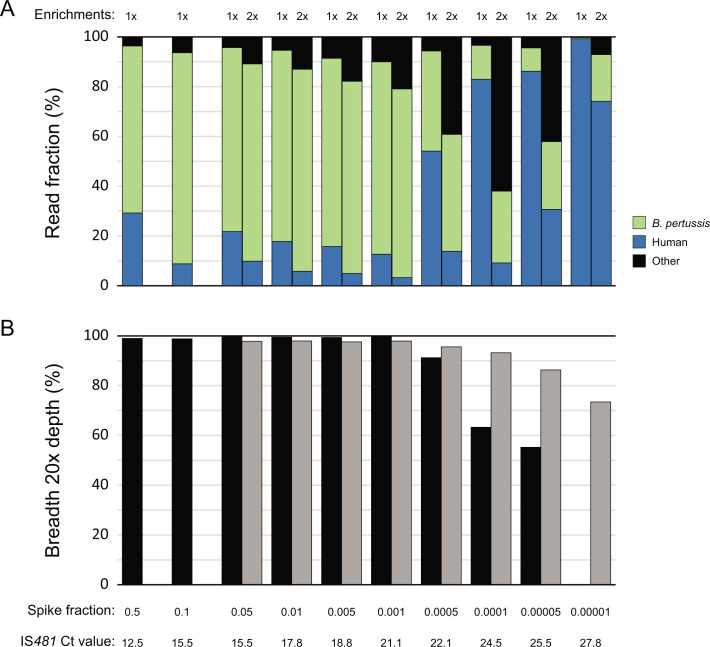
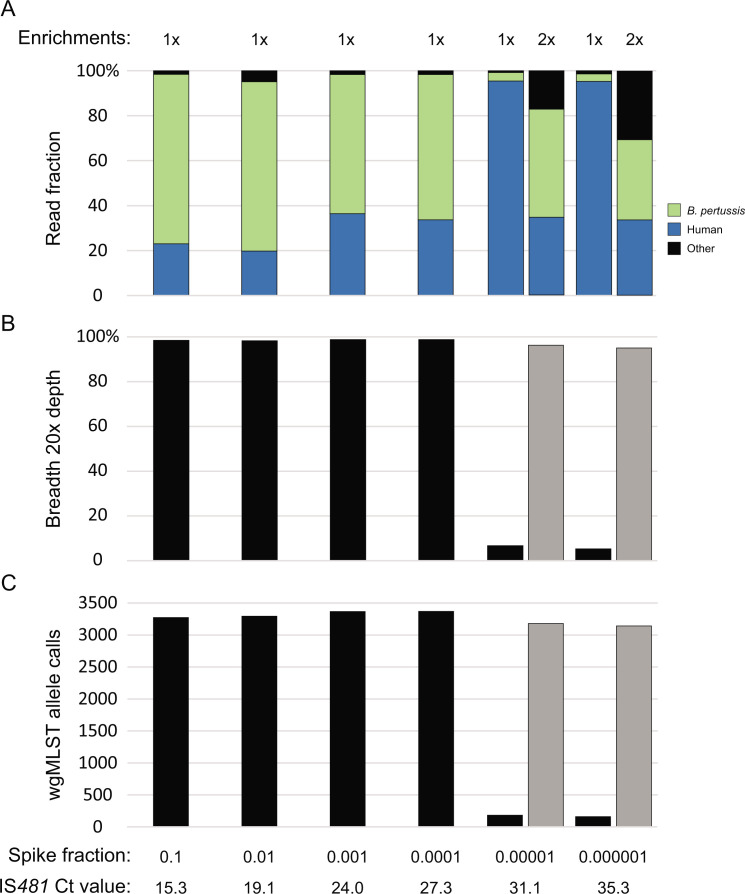
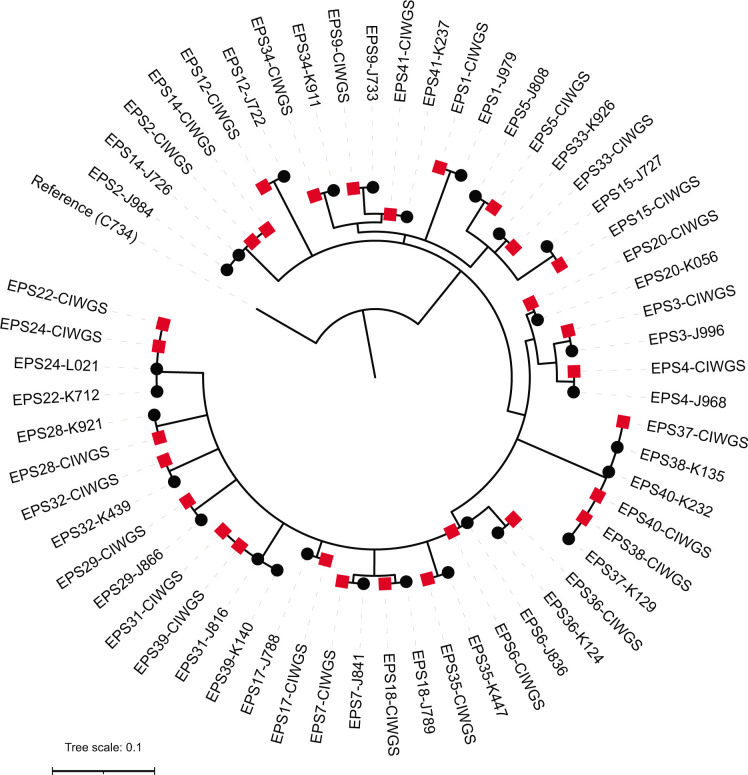
Whole-genome sequencing (WGS) of microbial pathogens recovered from patients with infectious disease facilitates high-resolution strain characterization and molecular epidemiology. However, increasing reliance on culture-independent methods to diagnose infectious diseases has resulted in few isolates available for WGS. Here, we report a novel culture-independent approach to genome characterization of Bordetella pertussis, the causative agent of pertussis and a paradigm for insufficient genomic surveillance due to limited culture of clinical isolates. Sequencing libraries constructed directly from residual pertussis-positive diagnostic nasopharyngeal specimens were hybridized with biotinylated RNA "baits" targeting B. pertussis fragments within complex mixtures that contained high concentrations of host and microbial background DNA. Recovery of B. pertussis genome sequence data was evaluated with mock and pooled negative clinical specimens spiked with reducing concentrations of either purified DNA or inactivated cells. Targeted enrichment increased the yield of B. pertussis sequencing reads up to 90% while simultaneously decreasing host reads to less than 10%. Filtered sequencing reads provided sufficient genome coverage to perform characterization via whole-genome single nucleotide polymorphisms and whole-genome multilocus sequencing typing. Moreover, these data were concordant with sequenced isolates recovered from the same specimens such that phylogenetic reconstructions from either consistently clustered the same putatively linked cases. The optimized protocol is suitable for nasopharyngeal specimens with diagnostic IS481 Ct < 35 and >10 ng DNA. Routine implementation of these methods could strengthen surveillance and study of pertussis resurgence by capturing additional cases with genomic characterization.
Keywords: Bordetella pertussis; metagenomics; surveillance; whole-genome sequencing.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Deciphering Bordetella pertussis epidemiology through culture-independent multiplex amplicon and metagenomic sequencing.J Clin Microbiol. 2024 Dec 11;62(12):e0117824. doi: 10.1128/jcm.01178-24. Epub 2024 Nov 4. J Clin Microbiol. 2024. PMID: 39494864 Free PMC article.
-
Genomic Sequencing of Bordetella pertussis for Epidemiology and Global Surveillance of Whooping Cough.Emerg Infect Dis. 2018 Jun;24(6):988-994. doi: 10.3201/eid2406.171464. Emerg Infect Dis. 2018. PMID: 29774847 Free PMC article.
-
Optimization of sample preparation for culture-independent sequencing of Bordetella pertussis.Microb Genom. 2020 Mar;6(3):e000332. doi: 10.1099/mgen.0.000332. Microb Genom. 2020. PMID: 32108565 Free PMC article.
-
A profile of the Simplexa™ Bordetella Direct assay for the detection and differentiation of Bordetella pertussis and Bordetella parapertussis in nasopharyngeal swabs.Expert Rev Mol Diagn. 2020 Sep;20(9):889-894. doi: 10.1080/14737159.2020.1819240. Epub 2020 Sep 20. Expert Rev Mol Diagn. 2020. PMID: 32885709 Review.
-
Bordetella pertussis epidemiology and evolution in the light of pertussis resurgence.Infect Genet Evol. 2016 Jun;40:136-143. doi: 10.1016/j.meegid.2016.02.032. Epub 2016 Feb 27. Infect Genet Evol. 2016. PMID: 26932577 Review.
Cited by
-
Deciphering Bordetella pertussis epidemiology through culture-independent multiplex amplicon and metagenomic sequencing.J Clin Microbiol. 2024 Dec 11;62(12):e0117824. doi: 10.1128/jcm.01178-24. Epub 2024 Nov 4. J Clin Microbiol. 2024. PMID: 39494864 Free PMC article.
-
Genomic reconstruction of Bacillus anthracis from complex environmental samples enables high-throughput identification and lineage assignment in Pakistan.Microb Genom. 2025 Jun;11(6):001422. doi: 10.1099/mgen.0.001422. Microb Genom. 2025. PMID: 40549420 Free PMC article.
-
Bordetella pertussis exhibits genomic diversity within patients and laboratory culture.mSystems. 2025 Jul 22;10(7):e0171724. doi: 10.1128/msystems.01717-24. Epub 2025 Jun 17. mSystems. 2025. PMID: 40525856 Free PMC article.
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
