

Prediction of protein-ligand binding affinity via deep learning models
- PMID: 38446737
- PMCID: PMC10939342
- DOI: 10.1093/bib/bbae081
Prediction of protein-ligand binding affinity via deep learning models
Erratum in
-
Correction to: Prediction of protein-ligand binding affinity via deep learning models.Brief Bioinform. 2024 May 23;25(4):bbae310. doi: 10.1093/bib/bbae310. Brief Bioinform. 2024. PMID: 38888458 Free PMC article. No abstract available.
Abstract
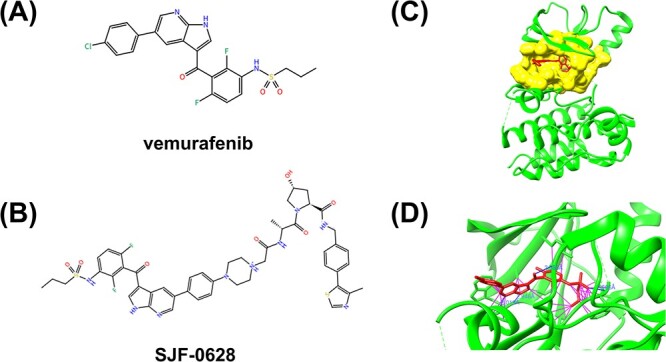
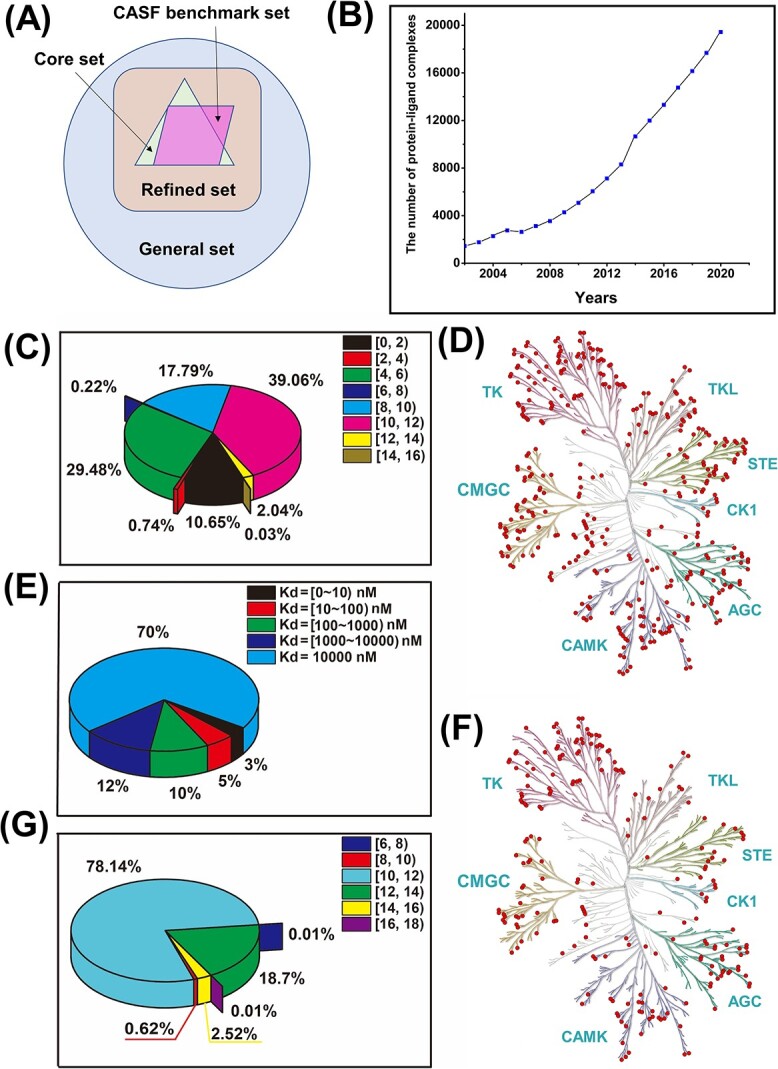
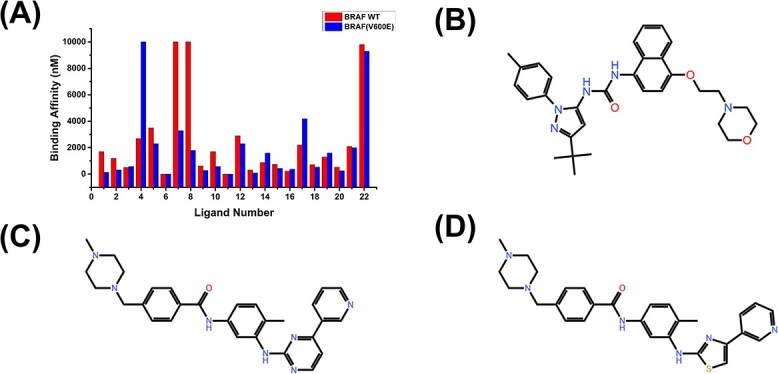
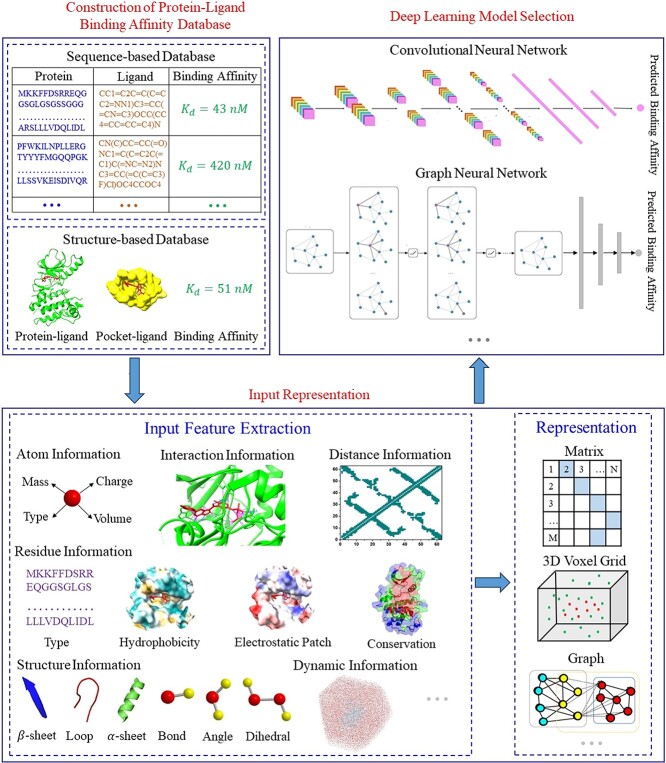
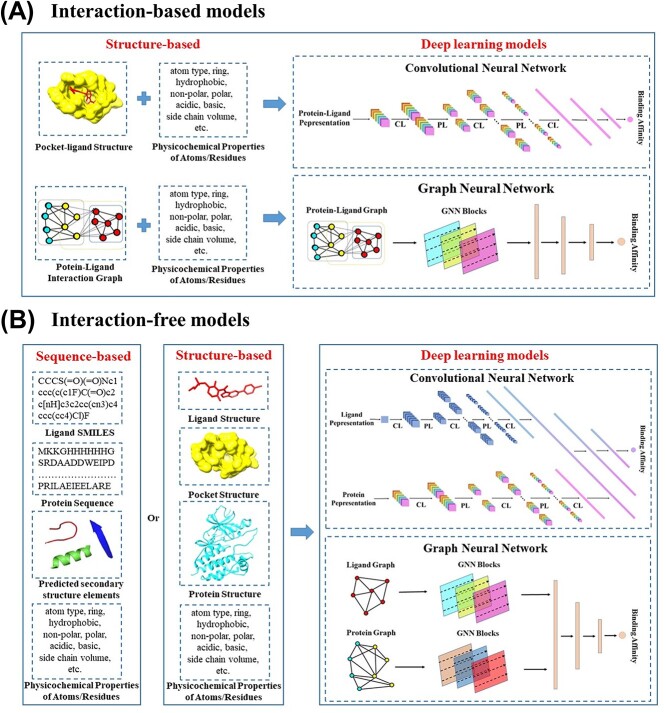
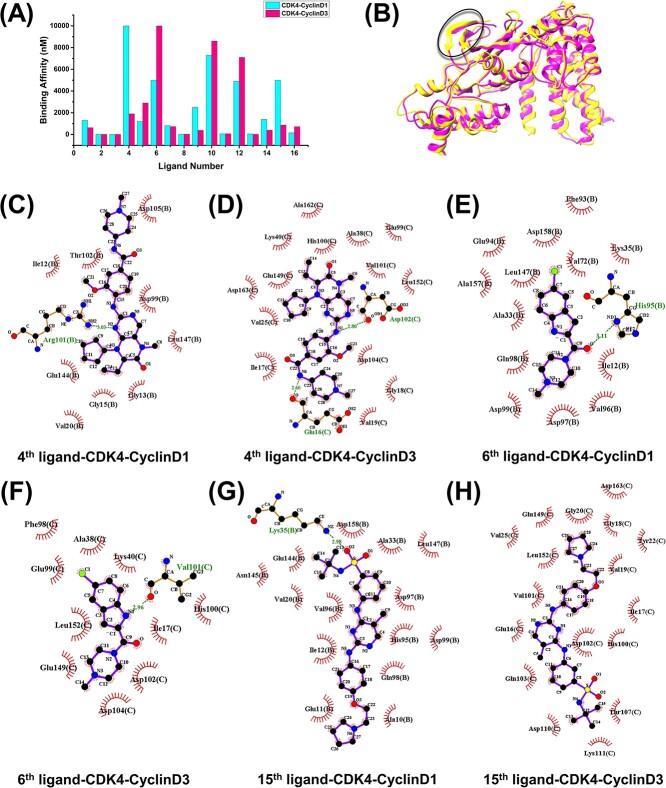
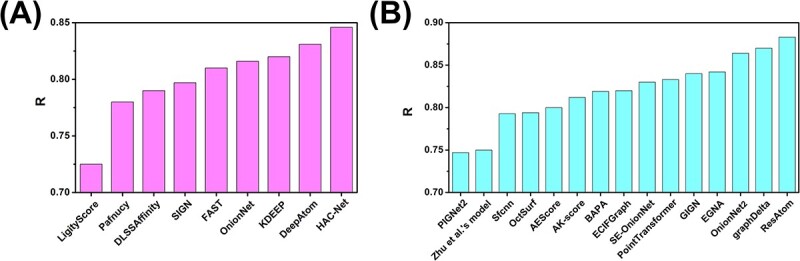
Accurately predicting the binding affinity between proteins and ligands is crucial in drug screening and optimization, but it is still a challenge in computer-aided drug design. The recent success of AlphaFold2 in predicting protein structures has brought new hope for deep learning (DL) models to accurately predict protein-ligand binding affinity. However, the current DL models still face limitations due to the low-quality database, inaccurate input representation and inappropriate model architecture. In this work, we review the computational methods, specifically DL-based models, used to predict protein-ligand binding affinity. We start with a brief introduction to protein-ligand binding affinity and the traditional computational methods used to calculate them. We then introduce the basic principles of DL models for predicting protein-ligand binding affinity. Next, we review the commonly used databases, input representations and DL models in this field. Finally, we discuss the potential challenges and future work in accurately predicting protein-ligand binding affinity via DL models.
Keywords: accurate prediction; database; deep learning model; input representation; protein–ligand binding affinity.
© The Author(s) 2024. Published by Oxford University Press.
Figures
Similar articles
-
Advances in Protein-Ligand Binding Affinity Prediction via Deep Learning: A Comprehensive Study of Datasets, Data Preprocessing Techniques, and Model Architectures.Curr Drug Targets. 2024;25(15):1041-1065. doi: 10.2174/0113894501330963240905083020. Curr Drug Targets. 2024. PMID: 39318214 Free PMC article. Review.
-
Advancing Ligand Docking through Deep Learning: Challenges and Prospects in Virtual Screening.Acc Chem Res. 2024 May 21;57(10):1500-1509. doi: 10.1021/acs.accounts.4c00093. Epub 2024 Apr 5. Acc Chem Res. 2024. PMID: 38577892 Review.
-
A New Hybrid Neural Network Deep Learning Method for Protein-Ligand Binding Affinity Prediction and De Novo Drug Design.Int J Mol Sci. 2022 Nov 11;23(22):13912. doi: 10.3390/ijms232213912. Int J Mol Sci. 2022. PMID: 36430386 Free PMC article.
-
DLSSAffinity: protein-ligand binding affinity prediction via a deep learning model.Phys Chem Chem Phys. 2022 May 4;24(17):10124-10133. doi: 10.1039/d1cp05558e. Phys Chem Chem Phys. 2022. PMID: 35416807
-
A new paradigm for applying deep learning to protein-ligand interaction prediction.Brief Bioinform. 2024 Mar 27;25(3):bbae145. doi: 10.1093/bib/bbae145. Brief Bioinform. 2024. PMID: 38581420 Free PMC article.
Cited by
-
PLAIG: Protein-Ligand Binding Affinity Prediction Using a Novel Interaction-Based Graph Neural Network Framework.ACS Bio Med Chem Au. 2025 Apr 29;5(3):447-463. doi: 10.1021/acsbiomedchemau.5c00053. eCollection 2025 Jun 18. ACS Bio Med Chem Au. 2025. PMID: 40556781 Free PMC article.
-
Deep Drug-Target Binding Affinity Prediction Base on Multiple Feature Extraction and Fusion.ACS Omega. 2025 Jan 10;10(2):2020-2032. doi: 10.1021/acsomega.4c08048. eCollection 2025 Jan 21. ACS Omega. 2025. PMID: 39866608 Free PMC article.
-
Edge-enhanced interaction graph network for protein-ligand binding affinity prediction.PLoS One. 2025 Apr 8;20(4):e0320465. doi: 10.1371/journal.pone.0320465. eCollection 2025. PLoS One. 2025. PMID: 40198678 Free PMC article.
-
DynamicDTA: Drug-Target Binding Affinity Prediction Using Dynamic Descriptors and Graph Representation.Interdiscip Sci. 2025 Jun 6. doi: 10.1007/s12539-025-00729-z. Online ahead of print. Interdiscip Sci. 2025. PMID: 40481301
-
Integrated modeling of protein and RNA.Brief Bioinform. 2024 Mar 27;25(3):bbae139. doi: 10.1093/bib/bbae139. Brief Bioinform. 2024. PMID: 38561980 Free PMC article. No abstract available.
References
-
- Volkamer A, Eid S, Turk S, et al. Pocketome of human kinases: prioritizing the ATP binding sites of (yet) untapped protein kinases for drug discovery. J Chem Inf Model 2015;55(3):538–49. - PubMed
