

Hepatic malonyl-CoA synthesis restrains gluconeogenesis by suppressing fat oxidation, pyruvate carboxylation, and amino acid availability
- PMID: 38447582
- PMCID: PMC11081827
- DOI: 10.1016/j.cmet.2024.02.004
Hepatic malonyl-CoA synthesis restrains gluconeogenesis by suppressing fat oxidation, pyruvate carboxylation, and amino acid availability
Abstract
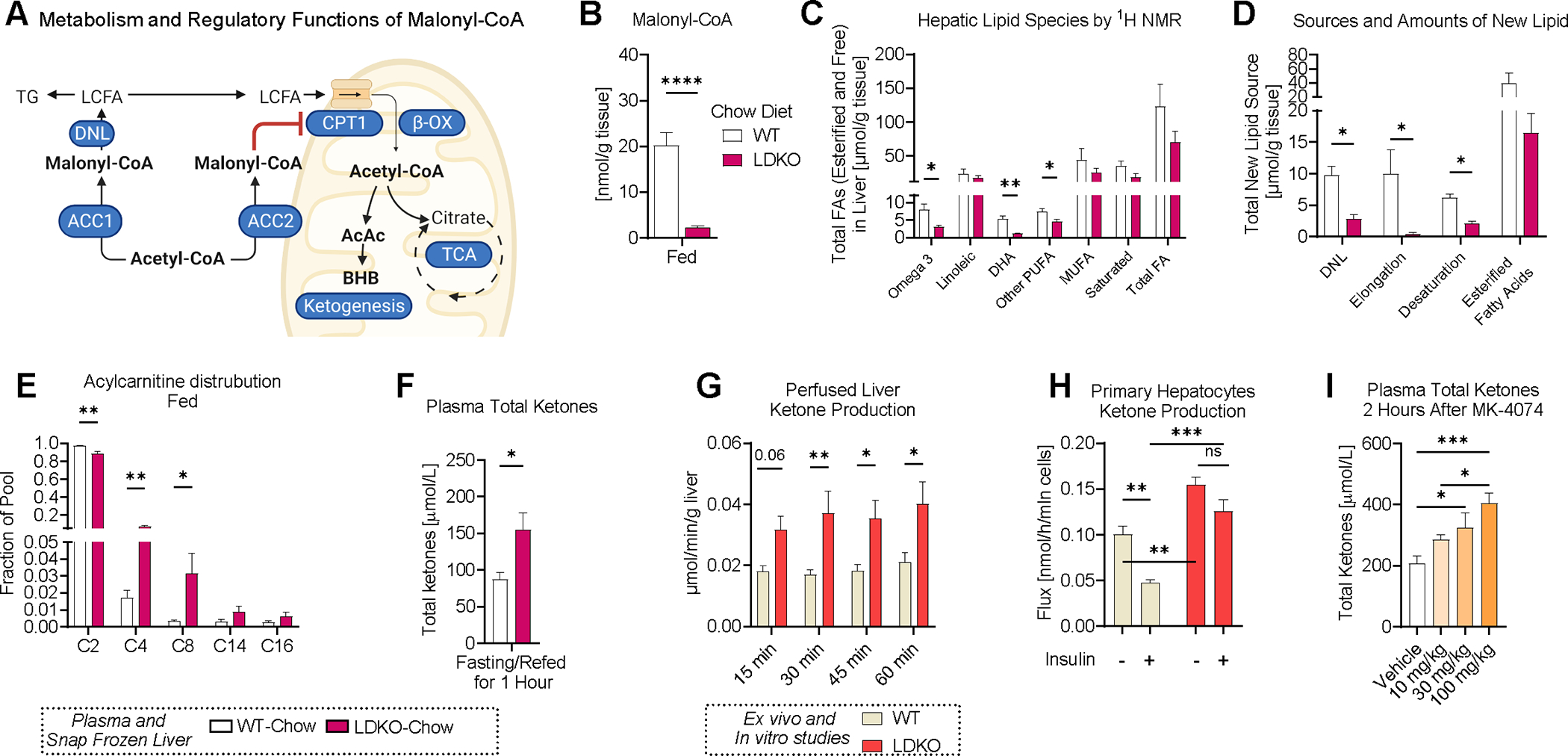
Acetyl-CoA carboxylase (ACC) promotes prandial liver metabolism by producing malonyl-CoA, a substrate for de novo lipogenesis and an inhibitor of CPT-1-mediated fat oxidation. We report that inhibition of ACC also produces unexpected secondary effects on metabolism. Liver-specific double ACC1/2 knockout (LDKO) or pharmacologic inhibition of ACC increased anaplerosis, tricarboxylic acid (TCA) cycle intermediates, and gluconeogenesis by activating hepatic CPT-1 and pyruvate carboxylase flux in the fed state. Fasting should have marginalized the role of ACC, but LDKO mice maintained elevated TCA cycle intermediates and preserved glycemia during fasting. These effects were accompanied by a compensatory induction of proteolysis and increased amino acid supply for gluconeogenesis, which was offset by increased protein synthesis during feeding. Such adaptations may be related to Nrf2 activity, which was induced by ACC inhibition and correlated with fasting amino acids. The findings reveal unexpected roles for malonyl-CoA synthesis in liver and provide insight into the broader effects of pharmacologic ACC inhibition.
Keywords: Nrf2; TCA cycle; acetyl-CoA carboxylase; anaplerosis; autophagy; gluconeogenesis; lipogenesis; malonyl-CoA; protein synthesis; proteolysis.
Copyright © 2024 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests J.D.H. is a consultant for Merck, Pfizer, and Regeneron.
Figures






References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
