

Cytidine deaminases APOBEC3C and APOBEC3D promote DNA replication stress resistance in pancreatic cancer cells
- PMID: 38448522
- PMCID: PMC12557688
- DOI: 10.1038/s43018-024-00742-z
Cytidine deaminases APOBEC3C and APOBEC3D promote DNA replication stress resistance in pancreatic cancer cells
Abstract
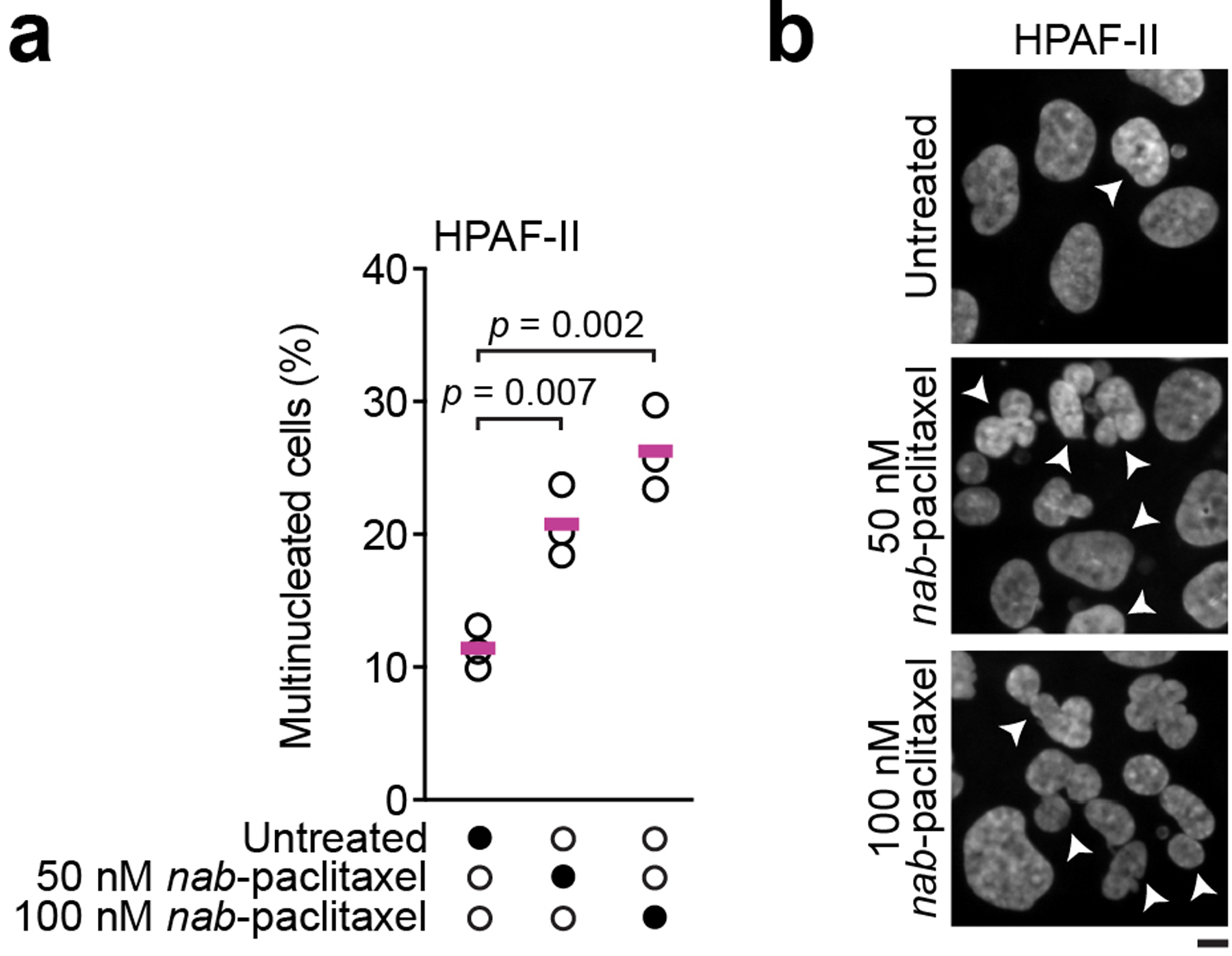
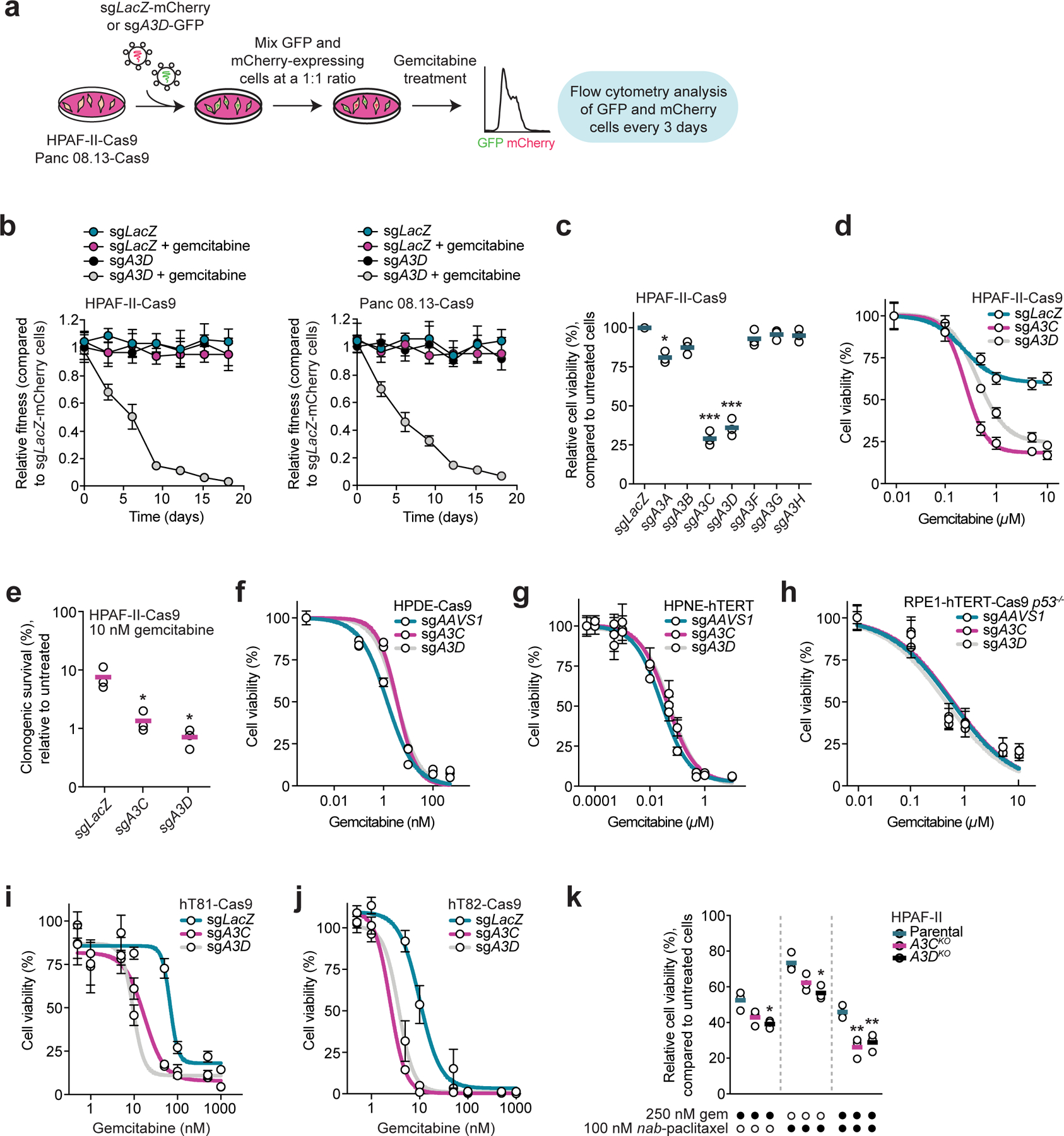
Gemcitabine is a potent inhibitor of DNA replication and is a mainstay therapeutic for diverse cancers, particularly pancreatic ductal adenocarcinoma (PDAC). However, most tumors remain refractory to gemcitabine therapies. Here, to define the cancer cell response to gemcitabine, we performed genome-scale CRISPR-Cas9 chemical-genetic screens in PDAC cells and found selective loss of cell fitness upon disruption of the cytidine deaminases APOBEC3C and APOBEC3D. Following gemcitabine treatment, APOBEC3C and APOBEC3D promote DNA replication stress resistance and cell survival by deaminating cytidines in the nuclear genome to ensure DNA replication fork restart and repair in PDAC cells. We provide evidence that the chemical-genetic interaction between APOBEC3C or APOBEC3D and gemcitabine is absent in nontransformed cells but is recapitulated across different PDAC cell lines, in PDAC organoids and in PDAC xenografts. Thus, we uncover roles for APOBEC3C and APOBEC3D in DNA replication stress resistance and offer plausible targets for improving gemcitabine-based therapies for PDAC.
© 2024. The Author(s), under exclusive licence to Springer Nature America, Inc.
Conflict of interest statement
DAT receives stock options from Leap Therapeutics, Surface Oncology, Sonata Therapeutics, and Mestag Therapeutics outside of the published work. DAT is scientific co-founder of Mestag Therapeutics. DAT has received research grant support from the Fibrogen, Mestag, and ONO Therapeutics. DAT receives grant funding from the Lustgarten Foundation, the NIH, and the Thompson Foundation. None of this work is related to the publication. TU and GWB were consultants for Repare Therapeutics. GWB received research support from Repare Therapeutics. No competing interests were reported by the remaining authors.
Figures














References
-
- Gorgoulis VG et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature 434, 907–913 (2005). - PubMed
-
- Bartkova J et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 434, 864–870 (2005). - PubMed
-
- Ubhi T & Brown GW Exploiting DNA replication stress for cancer treatment. Cancer Research 79, 1730–1739 (2019). - PubMed
-
- Hertel LW et al. Evaluation of the antitumor activity of gemcitabine (2’,2’-difluoro-2’-deoxycytidine). Cancer Research 50, 4417–22 (1990). - PubMed
Methods-only references
MeSH terms
Substances
Grants and funding
- PJT-463531/Gouvernement du Canada | Canadian Institutes of Health Research (Instituts de Recherche en Santé du Canada)
- R01 CA188134/CA/NCI NIH HHS/United States
- 5P30CA45508, U01CA210240, R01CA229699, U01CA224013, 1R01CA188134, and 1R01CA190092/U.S. Department of Health & Human Services | National Institutes of Health (NIH)
- R01 CA190092/CA/NCI NIH HHS/United States
- Innovate grant 706293/Canadian Cancer Society Research Institute (Société Canadienne du Cancer)
- U01 CA224013/CA/NCI NIH HHS/United States
- R01 CA229699/CA/NCI NIH HHS/United States
- grant 37660/Canada Foundation for Innovation (Fondation canadienne pour l'innovation)
- P30 CA045508/CA/NCI NIH HHS/United States
- U01 CA210240/CA/NCI NIH HHS/United States
- FDN-159913/Gouvernement du Canada | Canadian Institutes of Health Research (Instituts de Recherche en Santé du Canada)
- FRN 166094/Gouvernement du Canada | Canadian Institutes of Health Research (Instituts de Recherche en Santé du Canada)
- P.CTIP.1005/Ontario Institute for Cancer Research (Institut Ontarien de Recherche sur le Cancer)
- 552716/Simons Foundation
LinkOut - more resources
Full Text Sources
Medical
