

Disease trajectories in interstitial lung diseases - data from the EXCITING-ILD registry
- PMID: 38448953
- PMCID: PMC10919020
- DOI: 10.1186/s12931-024-02731-3
Disease trajectories in interstitial lung diseases - data from the EXCITING-ILD registry
Abstract
Background: Interstitial lung diseases (ILD) comprise a heterogeneous group of mainly chronic lung diseases with different disease trajectories. Progression (PF-ILD) occurs in up to 50% of patients and is associated with increased mortality.
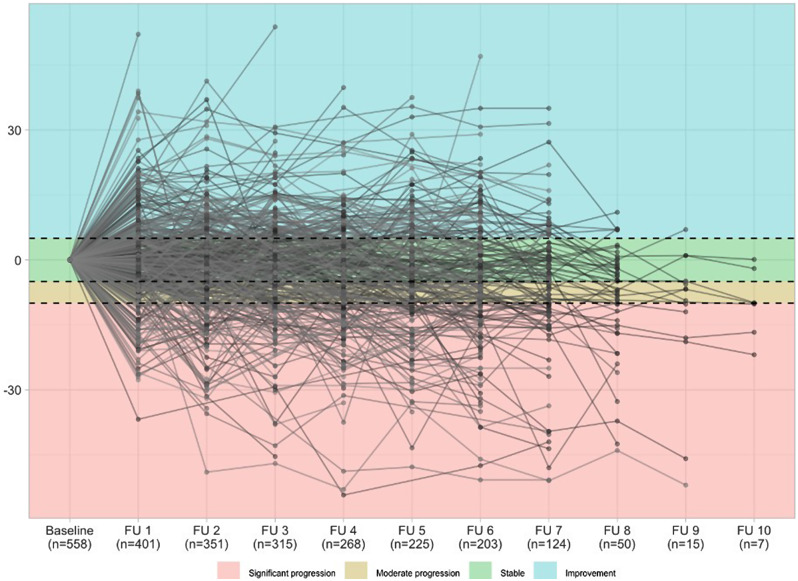
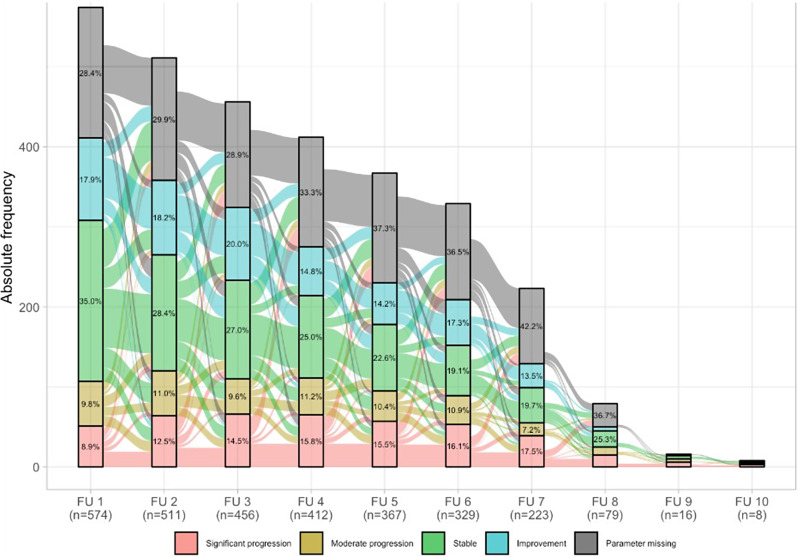
Methods: The EXCITING-ILD (Exploring Clinical and Epidemiological Characteristics of Interstitial Lung Diseases) registry was analysed for disease trajectories in different ILD. The course of disease was classified as significant (absolute forced vital capacity FVC decline > 10%) or moderate progression (FVC decline 5-10%), stable disease (FVC decline or increase < 5%) or improvement (FVC increase ≥ 5%) during time in registry. A second definition for PF-ILD included absolute decline in FVC % predicted ≥ 10% within 24 months or ≥ 1 respiratory-related hospitalisation. Risk factors for progression were determined by Cox proportional-hazard models and by logistic regression with forward selection. Kaplan-Meier curves were utilised to estimate survival time and time to progression.
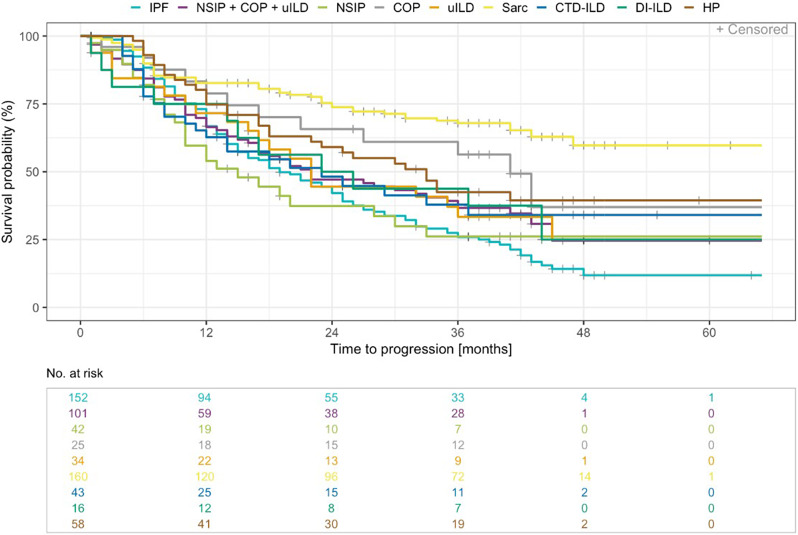
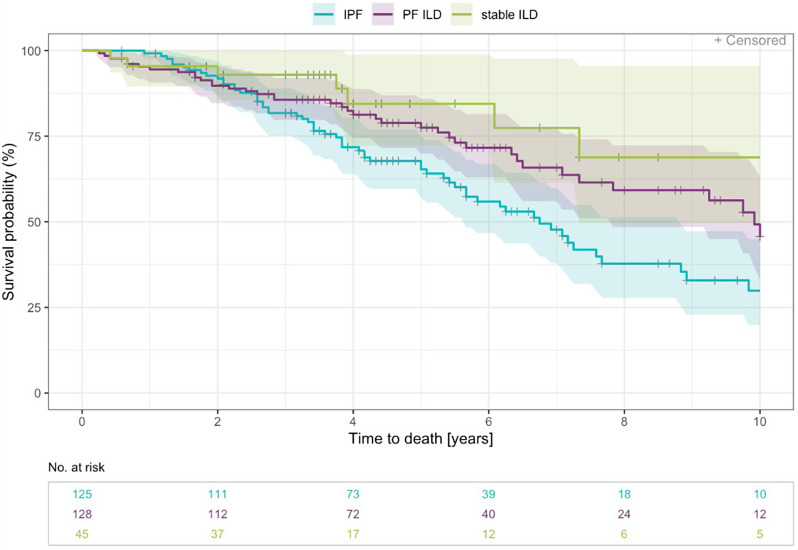
Results: Within the EXCITING-ILD registry 28.5% of the patients died (n = 171), mainly due to ILD (n = 71, 41.5%). Median survival time from date of diagnosis on was 15.5 years (range 0.1 to 34.4 years). From 601 included patients, progression was detected in 50.6% of the patients (n = 304) with shortest median time to progression in idiopathic NSIP (iNSIP; median 14.6 months) and idiopathic pulmonary fibrosis (IPF; median 18.9 months). Reasons for the determination as PF-ILD were mainly deterioration in lung function (PFT; 57.8%) and respiratory hospitalisations (40.6%). In multivariate analyses reduced baseline FVC together with age were significant predictors for progression (OR = 1.00, p < 0.001). Higher GAP indices were a significant risk factor for a shorter survival time (GAP stage III vs. I HR = 9.06, p < 0.001). A significant shorter survival time was found in IPF compared to sarcoidosis (HR = 0.04, p < 0.001), CTD-ILD (HR = 0.33, p < 0.001), and HP (HR = 0.30, p < 0.001). Patients with at least one reported ILD exacerbation as a reason for hospitalisation had a median survival time of 7.3 years (range 0.1 to 34.4 years) compared to 19.6 years (range 0.3 to 19.6 years) in patients without exacerbations (HR = 0.39, p < 0.001).
Conclusion: Disease progression is common in all ILD and associated with increased mortality. Most important risk factors for progression are impaired baseline forced vital capacity and higher age, as well as acute exacerbations and respiratory hospitalisations for mortality. Early detection of progression remains challenging, further clinical criteria in addition to PFT might be helpful.
Keywords: ILD; IPF; Mortality; Progression; Risk factors.
© 2024. The Author(s).
Conflict of interest statement
KB received payment for lectures from Boehringer Ingelheim and a grant from Sarkoidose-Netzwerk e.V. LH received fees for consulting and lectures from Boehringer-Ingelheim, GSK, AstraZeneca, Pfizer and BMS. PH received fees for lectures from Galapagos, GSK, Boehringer Ingelheim, AstraZeneca, Roche. DS reports fees for lectures or consultations from AstraZeneca, Boehringer Ingelheim, Chiesi, GSK, Janssen, MSD, Sanofi, all outside the submitted work. CS reports fees for lectures GSK, AstraZeneca, Boehringer Ingelheim, Berlin Chemie. MJ reports fees for lectures or consultations from AstraZeneca, Bencard, Boehringer Ingelheim, GSK, HAL Allergy, Sanofi, all outside the submitted work. SV reports fees from Berlin Chemie, GSK, Lilly, Pfizer, and Boehringer-Ingelheim. LS reports consulting fees from Galapagos. JB reports personal fees for lectures and consulting from Astra-Zeneca, Biogen, Boehringer-Ingelheim, BMS, Ferrer, Novartis, Roche, and Sanofi. AG reports grants, leture payments and/or consulting fees from Boehringer Ingelheim, Roche, Lung Therapeutics and Pieris. MP has received payment or honoraria for lectures, presentations or educational events from Boehringer Ingelheim and AstraZeneca. VS received support for attending meetings from CSL Behring. PM received fees for consulting and lectures from Boehringer-Ingelheim and Roche. MK reports grants, consulting fees, or payment for lectures from Boehringer Ingelheim, Galapagos, AstraZeneca, BMS and Roche. All other authors have nothing to disclose. The registry was supported by an unrestricted grant by Roche.
The authors declare no competing interests.
Figures
References
-
- Valeyre D, Duchemann B, Annesi-Maesano I et al. Interstitial lung diseases, in Respiratory Epidemiology, T. Welte, I. Annesi-Maesano, G. Viegi, and B. Lundbäck,Eds., vol. 65 of ERSMonograph, Chap. 6, ERS, 2014.
-
- American Thoracic Society and European Respiratory Society American Thoracic Society/European Respiratory Society international multidisciplinary consensus classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med. 2002;165(2):277–304. doi: 10.1164/ajrccm.165.2.ats01. - DOI - PubMed
-
- Travis WD, Costabel U, Hansell D. An official American Thoracic Society/European Respiratory Society statement: update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med. 2013;188(6):733–48. doi: 10.1164/rccm.201308-1483ST. - DOI - PMC - PubMed
-
- Raghu G, Remy-Jardin M, Richeldi L, Thomson CC, Inoue Y, Johkoh T, Kreuter M, Lynch DA, Maher TM, Martinez FJ, Molina-Molina M, Myers JL, Nicholson AG, Ryerson CJ, Strek ME, Troy LK, Wijsenbeek M, Mammen MJ, Hossain T, Bissell BD, Herman DD, Hon SM, Kheir F, Khor YH, Macrea M, Antoniou KM, Bouros D, Buendia-Roldan I, Caro F, Crestani B, Ho L, Morisset J, Olson AL, Podolanczuk A, Poletti V, Selman M, Ewing T, Jones S, Knight SL, Ghazipura M, Wilson KC. Idiopathic pulmonary fibrosis (an update) and progressive pulmonary fibrosis in adults: an Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am J Respir Crit Care Med. 2022;205(9):e18–e47. doi: 10.1164/rccm.202202-0399ST. - DOI - PMC - PubMed
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
