

Mito-metformin protects against mitochondrial dysfunction and dopaminergic neuronal degeneration by activating upstream PKD1 signaling in cell culture and MitoPark animal models of Parkinson's disease
- PMID: 38449738
- PMCID: PMC10915001
- DOI: 10.3389/fnins.2024.1356703
Mito-metformin protects against mitochondrial dysfunction and dopaminergic neuronal degeneration by activating upstream PKD1 signaling in cell culture and MitoPark animal models of Parkinson's disease
Abstract
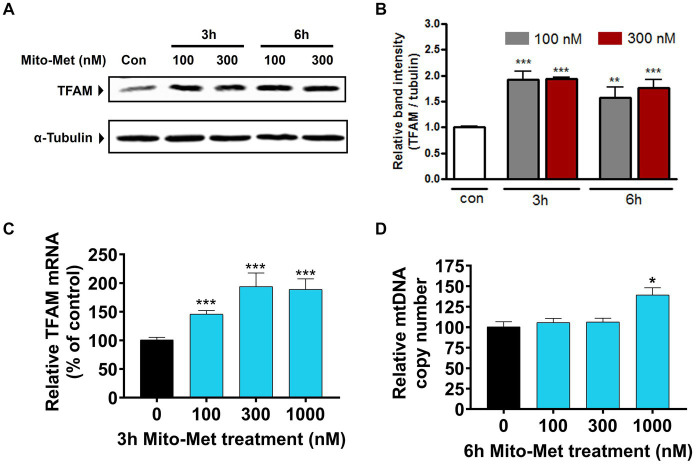
Impaired mitochondrial function and biogenesis have strongly been implicated in the pathogenesis of Parkinson's disease (PD). Thus, identifying the key signaling mechanisms regulating mitochondrial biogenesis is crucial to developing new treatment strategies for PD. We previously reported that protein kinase D1 (PKD1) activation protects against neuronal cell death in PD models by regulating mitochondrial biogenesis. To further harness the translational drug discovery potential of targeting PKD1-mediated neuroprotective signaling, we synthesized mito-metformin (Mito-Met), a mitochondria-targeted analog derived from conjugating the anti-diabetic drug metformin with a triphenylphosphonium functional group, and then evaluated the preclinical efficacy of Mito-Met in cell culture and MitoPark animal models of PD. Mito-Met (100-300 nM) significantly activated PKD1 phosphorylation, as well as downstream Akt and AMPKα phosphorylation, more potently than metformin, in N27 dopaminergic neuronal cells. Furthermore, treatment with Mito-Met upregulated the mRNA and protein expression of mitochondrial transcription factor A (TFAM) implying that Mito-Met can promote mitochondrial biogenesis. Interestingly, Mito-Met significantly increased mitochondrial bioenergetics capacity in N27 dopaminergic cells. Mito-Met also reduced mitochondrial fragmentation induced by the Parkinsonian neurotoxicant MPP+ in N27 cells and protected against MPP+-induced TH-positive neurite loss in primary neurons. More importantly, Mito-Met treatment (10 mg/kg, oral gavage for 8 week) significantly improved motor deficits and reduced striatal dopamine depletion in MitoPark mice. Taken together, our results demonstrate that Mito-Met possesses profound neuroprotective effects in both in vitro and in vivo models of PD, suggesting that pharmacological activation of PKD1 signaling could be a novel neuroprotective translational strategy in PD and other related neurocognitive diseases.
Keywords: MitoPark; PKD1; Parkinson’s disease; metformin; mitochondria; mitochondrial biogenesis; neuroprotection.
Copyright © 2024 Ay, Charli, Langley, Jang, Padhi, Jin, Anantharam, Kalyanaraman, Kanthasamy and Kanthasamy.
Conflict of interest statement
AnK and VA have an equity interest in PK Biosciences Corporation and Probiome Therapeutics, located at the University of Georgia, Athens, GA. The terms of this arrangement have been reviewed and approved by the University of Georgia in accordance with their conflict-of-interest policies. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures







References
-
- Asaithambi A., Kanthasamy A., Saminathan H., Anantharam V., Kanthasamy A. G. (2011). Protein kinase D1 (PKD1) activation mediates a compensatory protective response during early stages of oxidative stress-induced neuronal degeneration. Mol. Neurodegener. 6:43. doi: 10.1186/1750-1326-6-43, PMID: - DOI - PMC - PubMed
-
- Ay M., Jin H., Harischandra D. S., Asaithambi A., Kanthasamy A., Anantharam V., et al. (2015). Molecular cloning, epigenetic regulation, and functional characterization of Prkd1 gene promoter in dopaminergic cell culture models of Parkinson's disease. J. Neurochem. 135, 402–415. doi: 10.1111/jnc.13261, PMID: - DOI - PMC - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous