

Targeting CCL2/CCR2 Signaling Overcomes MEK Inhibitor Resistance in Acute Myeloid Leukemia
- PMID: 38451486
- PMCID: PMC11094423
- DOI: 10.1158/1078-0432.CCR-23-2654
Targeting CCL2/CCR2 Signaling Overcomes MEK Inhibitor Resistance in Acute Myeloid Leukemia
Abstract
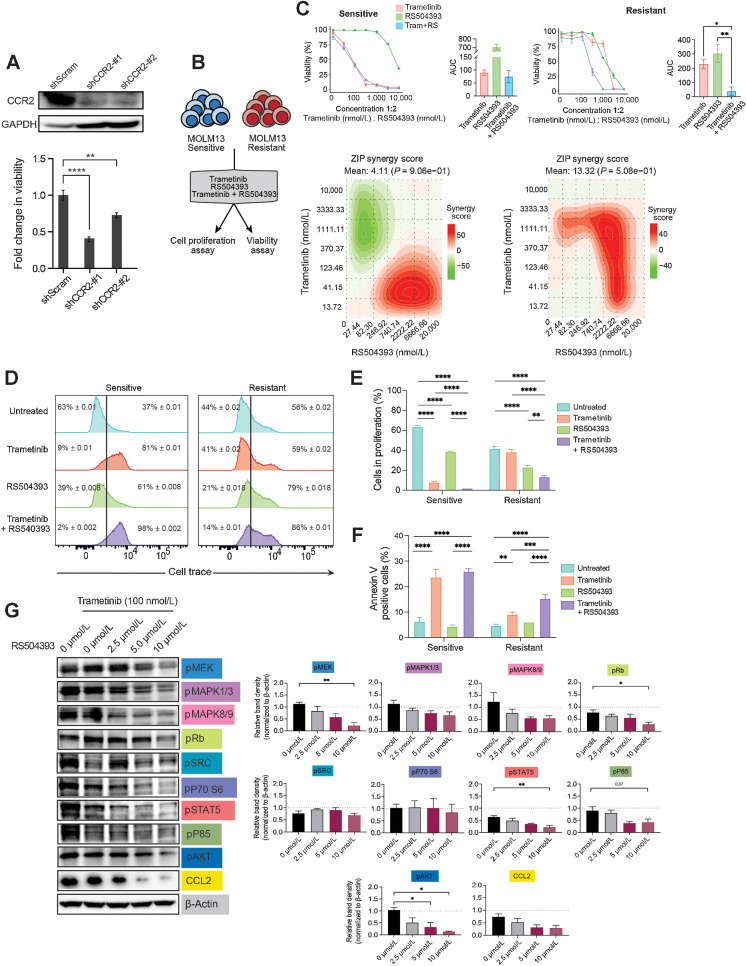
Purpose: Emerging evidence underscores the critical role of extrinsic factors within the microenvironment in protecting leukemia cells from therapeutic interventions, driving disease progression, and promoting drug resistance in acute myeloid leukemia (AML). This finding emphasizes the need for the identification of targeted therapies that inhibit intrinsic and extrinsic signaling to overcome drug resistance in AML.
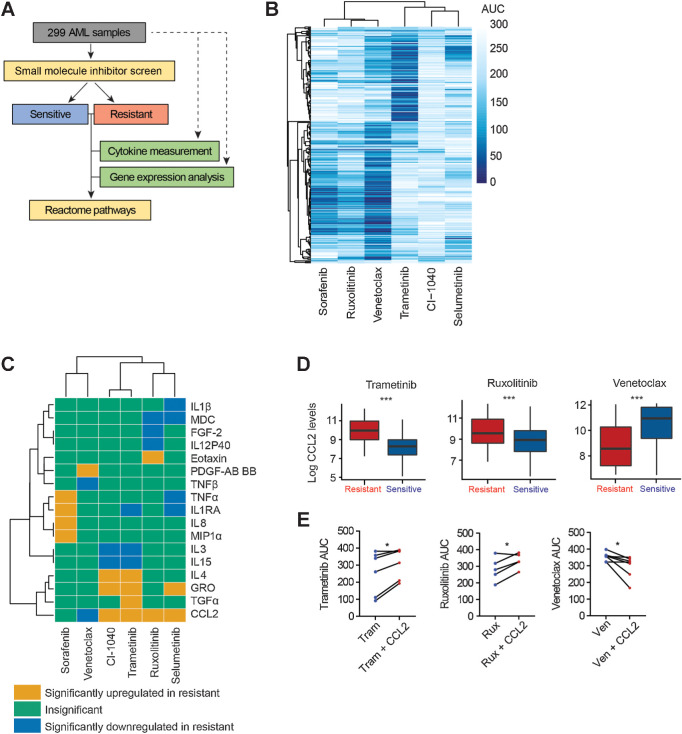
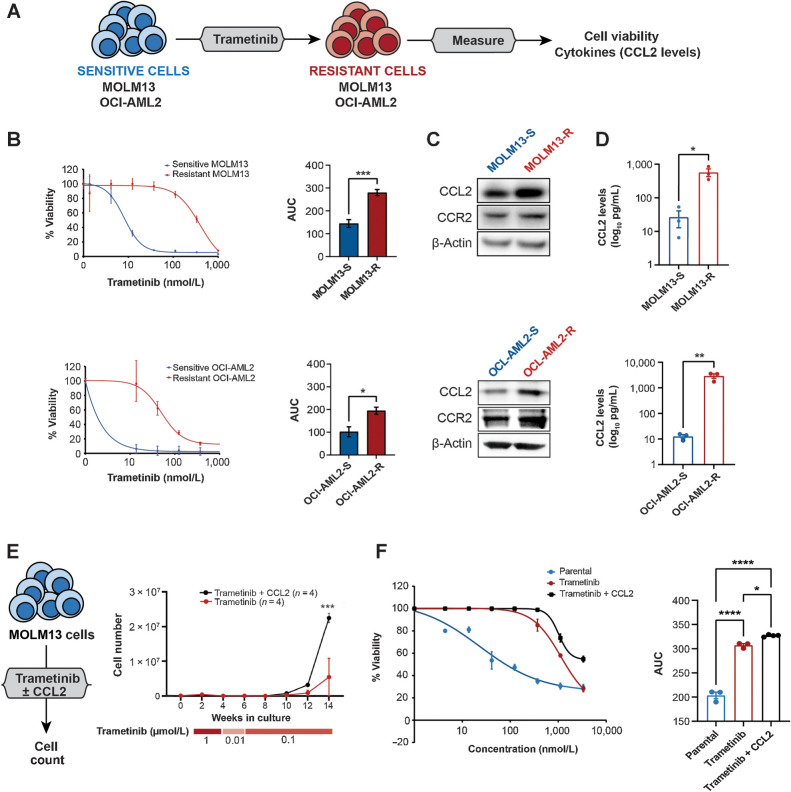
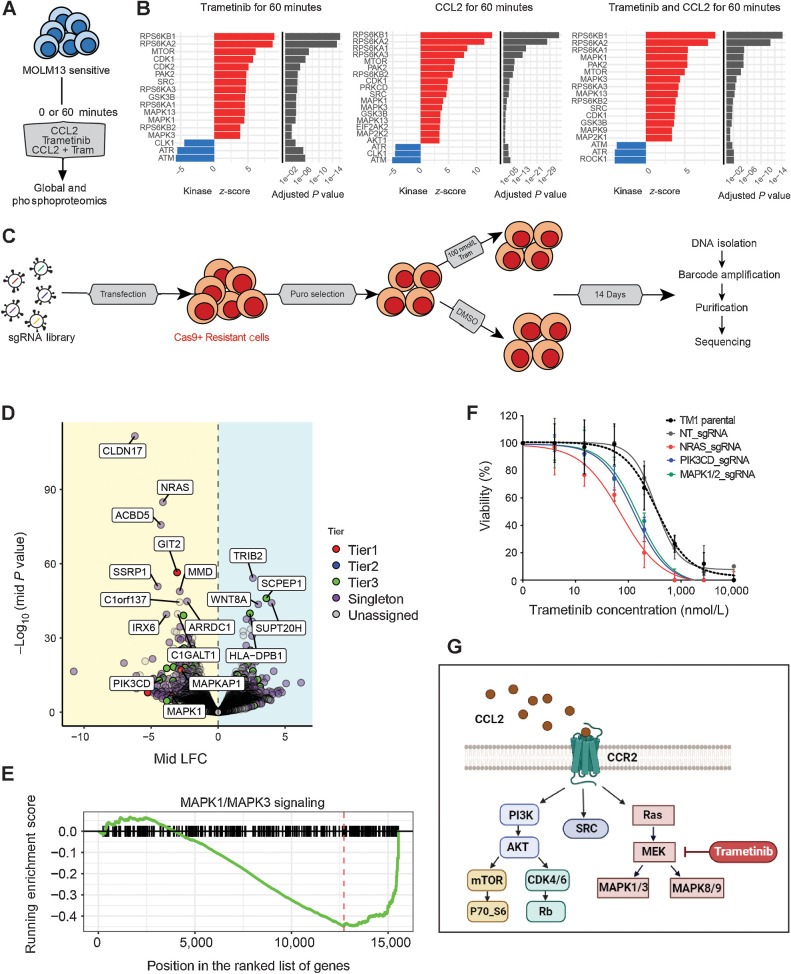
Experimental design: We performed a comprehensive analysis utilizing a cohort of ∼300 AML patient samples. This analysis encompassed the evaluation of secreted cytokines/growth factors, gene expression, and ex vivo drug sensitivity to small molecules. Our investigation pinpointed a notable association between elevated levels of CCL2 and diminished sensitivity to the MEK inhibitors (MEKi). We validated this association through loss-of-function and pharmacologic inhibition studies. Further, we deployed global phosphoproteomics and CRISPR/Cas9 screening to identify the mechanism of CCR2-mediated MEKi resistance in AML.
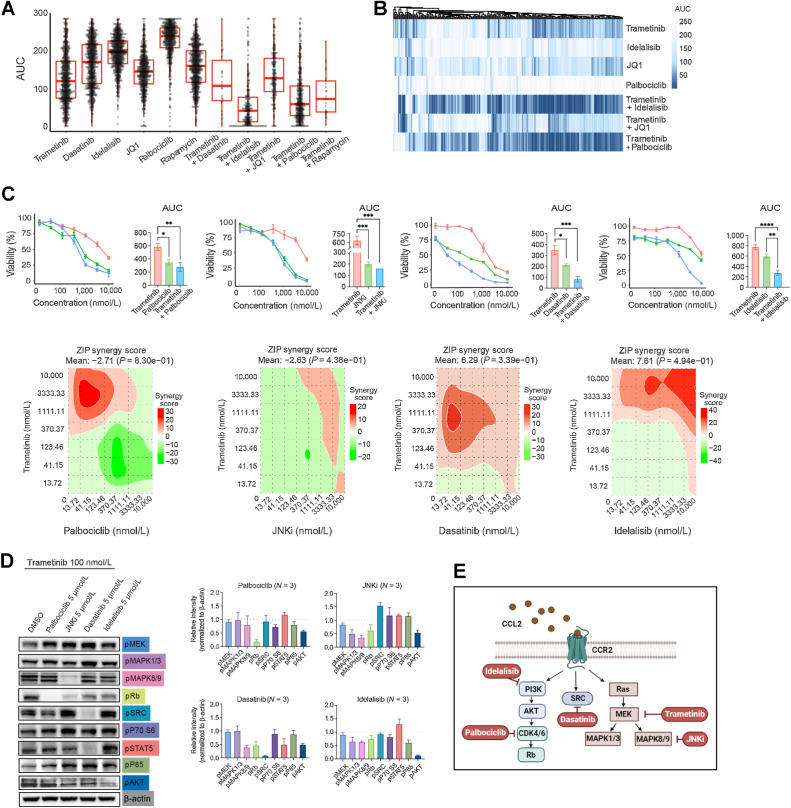
Results: Our multifaceted analysis unveiled that CCL2 activates multiple prosurvival pathways, including MAPK and cell-cycle regulation in MEKi-resistant cells. Employing combination strategies to simultaneously target these pathways heightened growth inhibition in AML cells. Both genetic and pharmacologic inhibition of CCR2 sensitized AML cells to trametinib, suppressing proliferation while enhancing apoptosis. These findings underscore a new role for CCL2 in MEKi resistance, offering combination therapies as an avenue to circumvent this resistance.
Conclusions: Our study demonstrates a compelling rationale for translating CCL2/CCR2 axis inhibitors in combination with MEK pathway-targeting therapies, as a potent strategy for combating drug resistance in AML. This approach has the potential to enhance the efficacy of treatments to improve AML patient outcomes.
©2024 The Authors; Published by the American Association for Cancer Research.
Figures
References
-
- 2018 National Cancer Institute: Surveillance, Epidemiology, and End Results Program. Available from: https://seer.cancer.gov/statfacts/.
-
- Jemal A, Siegel R, Xu J, Ward E. Cancer statistics 2010. CA Cancer J Clin 2010;60:277–300. - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
